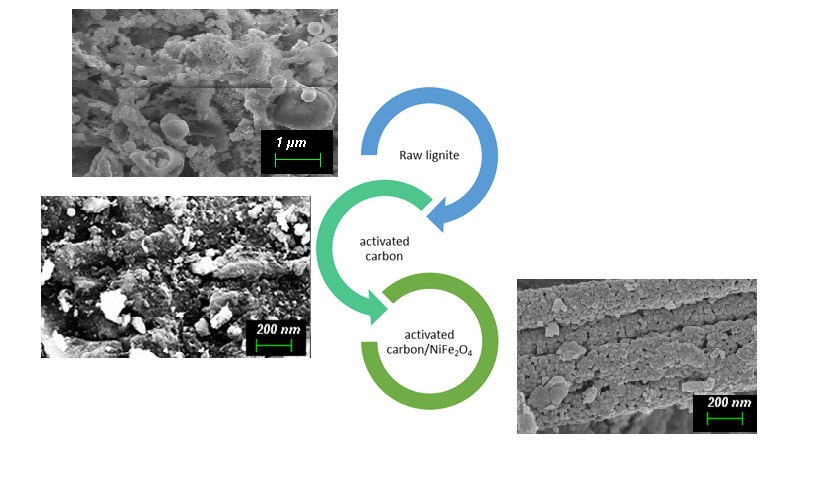
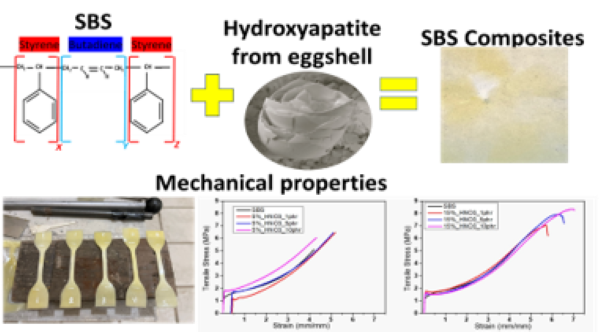
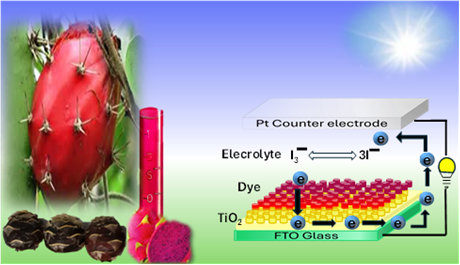
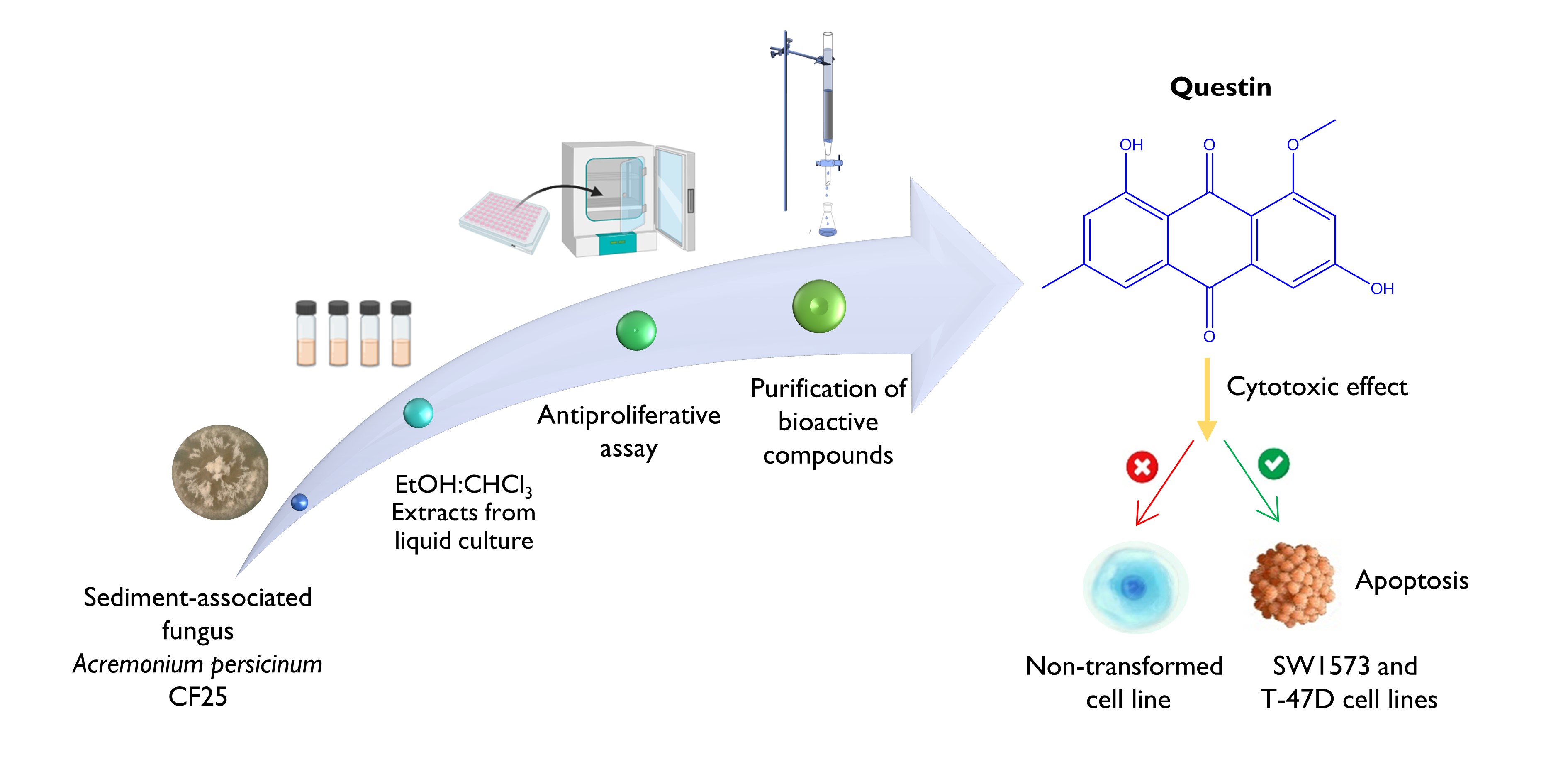
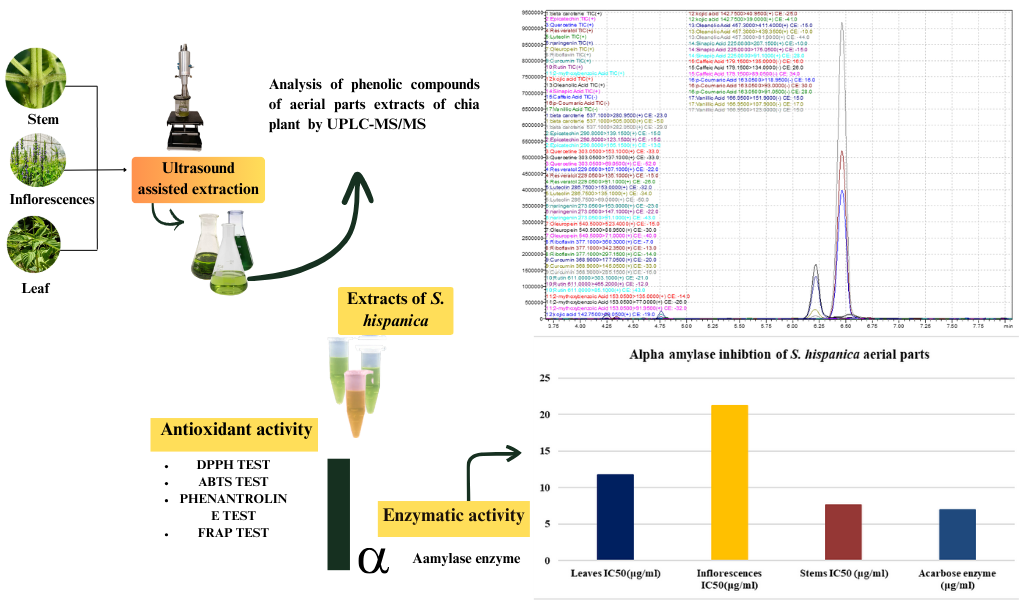
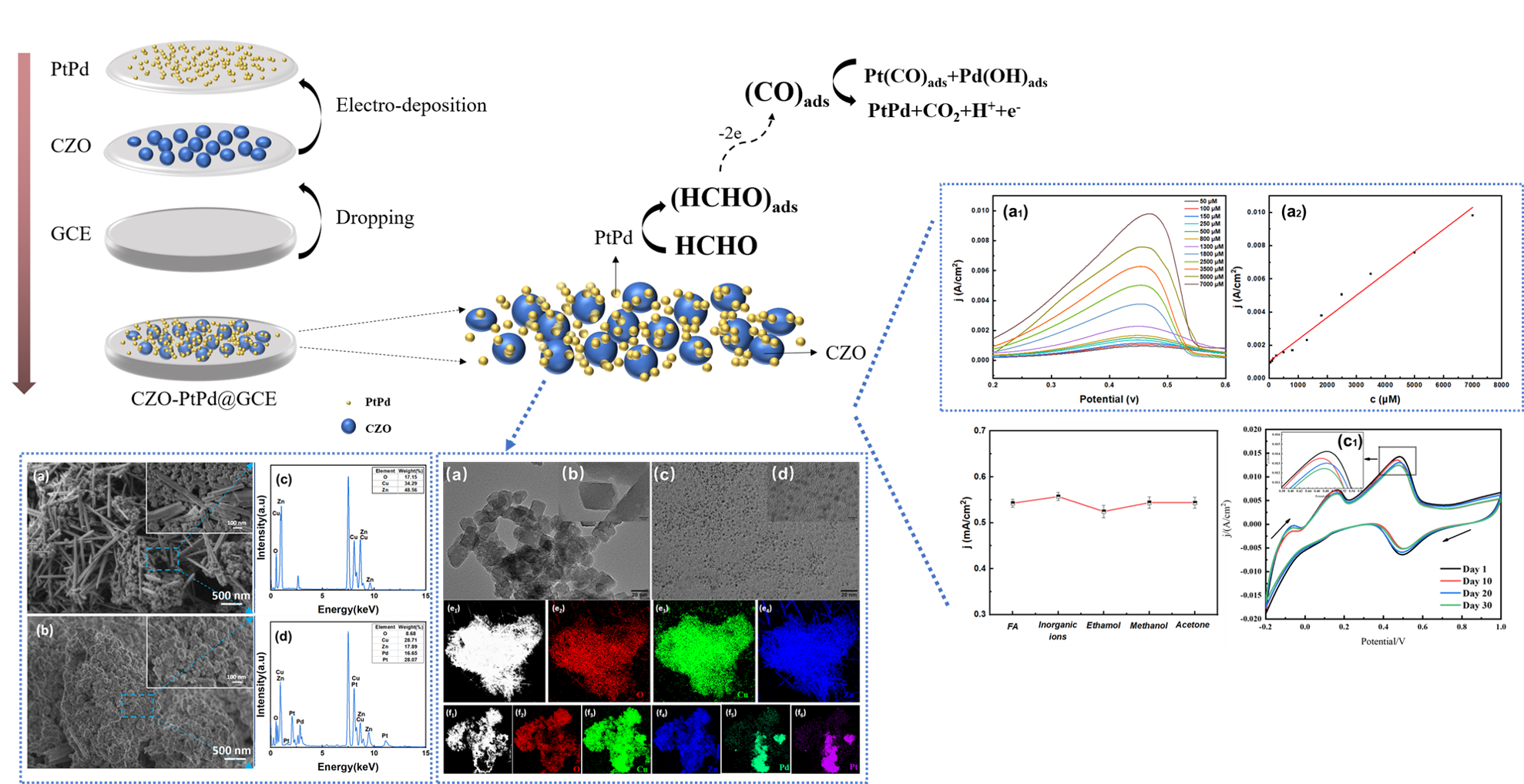
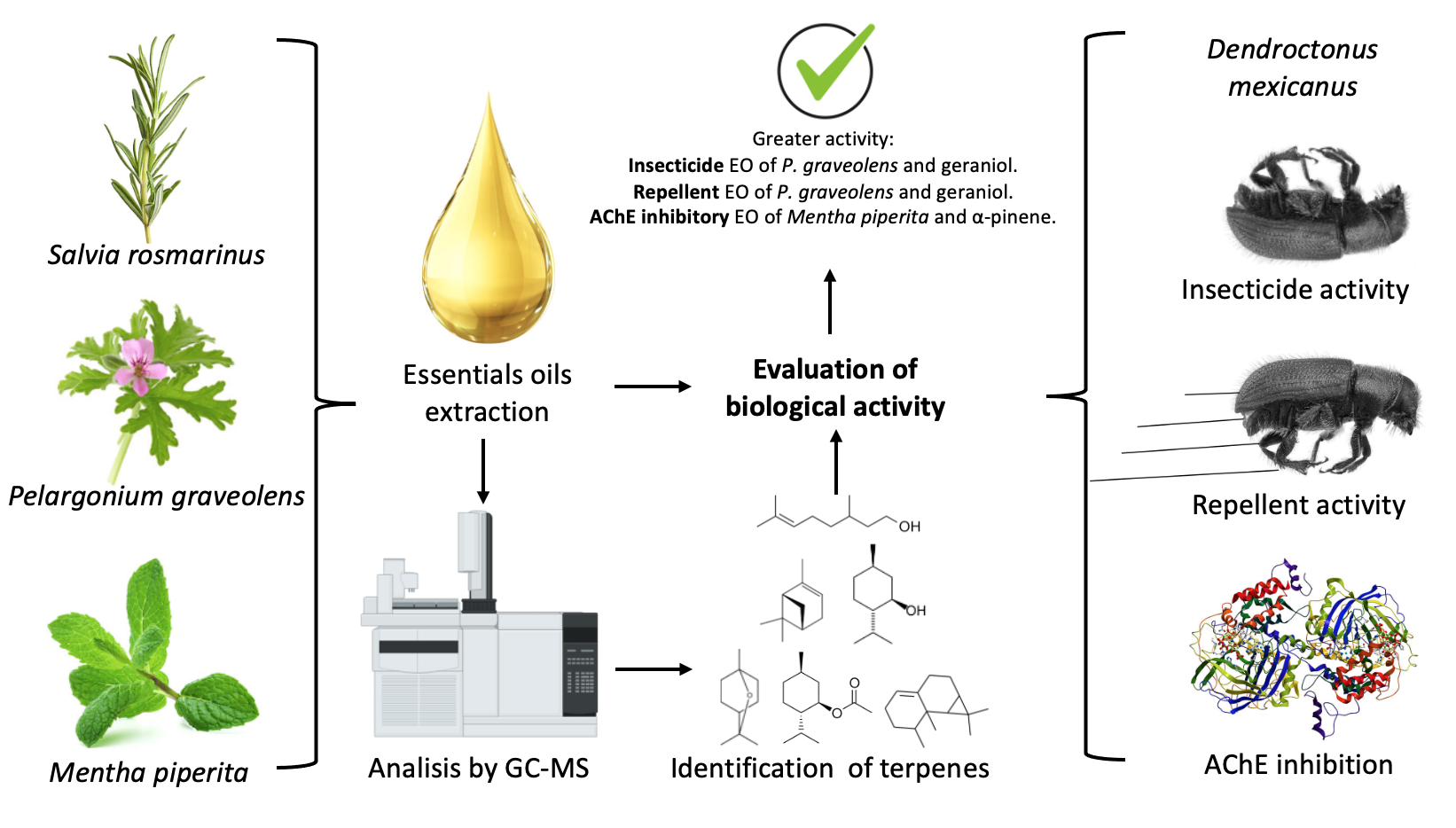
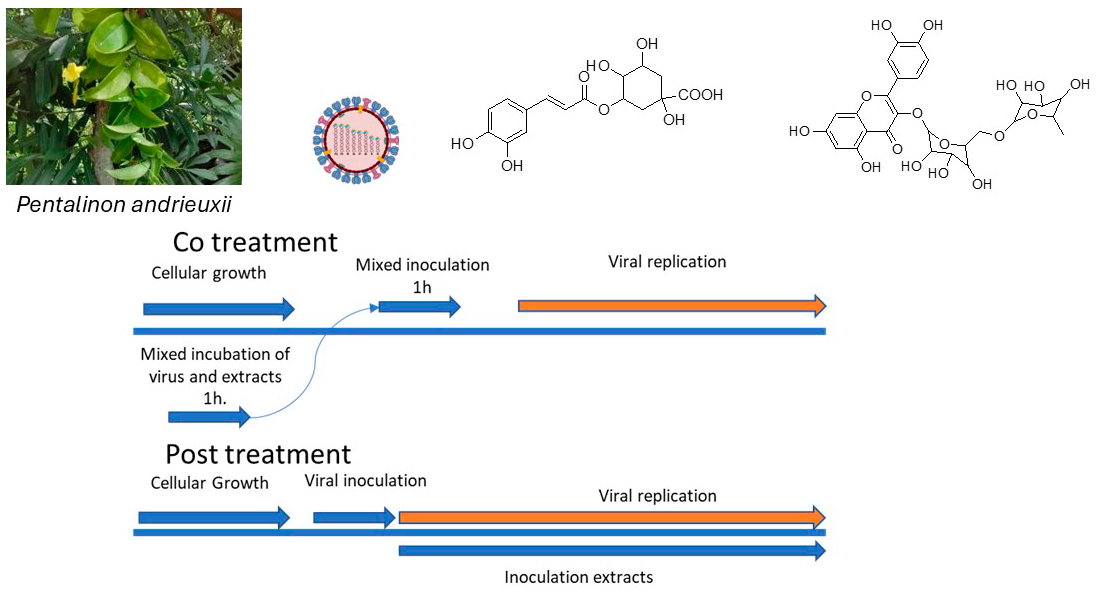
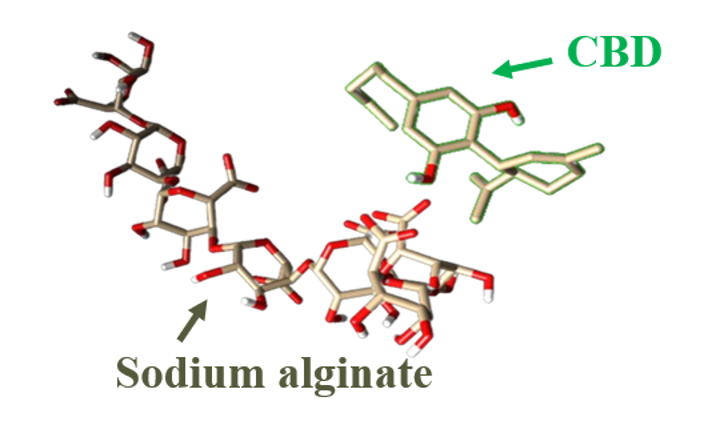
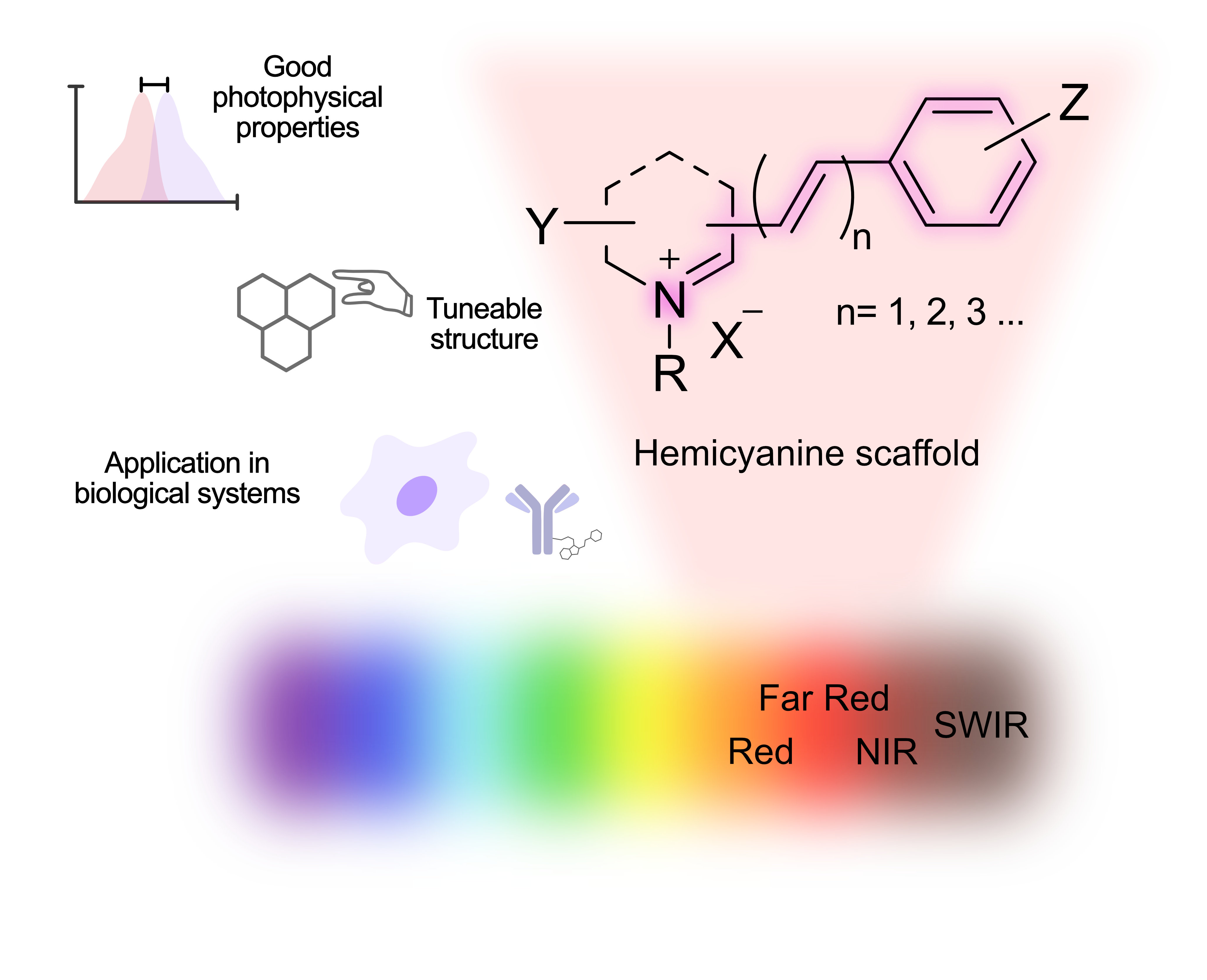
The Journal of the Mexican Chemical Society (J. Mex. Chem. Soc.) is a scientific, blind, peer-reviewed, open-access publication covering all areas of chemistry and its sub-disciplines (i.e. medicinal chemistry, natural products, electrochemistry, material science, computational chemistry, organic chemistry, bioinorganic chemistry, etc.). It is devoted to facilitating the worldwide advancement of our understanding of chemistry. It will primarily publish original contributions of research in all branches of the theory and practice of chemistry in its broadest context, as well as critical reviews in active areas of chemical research where the author has published significant contributions.
Starting 2026, The J. Mex. Chem. Soc. (JMCS) operates as a Scientific Continuous Publication (CP), in which the language of submission and publication is English. To be suitable for publication in J. Mex. Chem. Soc., manuscripts must describe novel aspects of chemistry, high-quality results, and discussion, with excellent bibliographic support, and contribute to the development of the field. Routine or incremental work is not suitable for publication in J. Mex. Chem. Soc. Authors are encouraged to send contributions in electronic form. Our online submission system (OJS) guides you stepwise through the process of entering your article details and uploading your files to the journal´s platform.
Article Processing Charges (APCs): From February 2026, the J. Mex. Chem. Soc. will charge upon acceptance the amount of $5,250.00 MXN or $3,150.00 MXN (to SQM active members) only for submissions received from January 1st 2026 onwards. Accepted submissions received during 2025 will not be charged according to the 2025 guidelines.
Special Issues: In 2026, the J. Mex. Chem. Soc will neither publish nor accept applications for Special Issues until further notice.
• Article types
1. Research Papers (Full paper): Should be complete and authoritative accounts of work that has special significance and general interest, presented clearly and concisely.
2. Critical Review Papers: In active areas of chemical research where the author has published significant contributions. The editorial board should invite the authors to submit this paper type.
| JIF 2025 | |
| 3 years | 0.8 |
| 5 years | 1.0 |