Journal of the Mexican Chemical Society, vol. 65, no. 1, 2021
Sociedad Química de México A.C.
Rafael Hernández-Tenorio
Universidad Autónoma de Nuevo León, Mexico
Jorge Luis Guzmán-Mar
Universidad Autónoma de Nuevo León, Mexico
Laura Hinojosa-Reyes
Universidad Autónoma de Nuevo León, Mexico
Norma Ramos-Delgado
Instituto Tecnológico de Nuevo Léon, Mexico
Aracely Hernández-Ramírez *
Universidad Autónoma de Nuevo León, Mexico
Received: 05 September 2020
Accepted: 16 October 2020
Funding
Funding source: Consejo Nacional de Ciencia y Tecnología (CONACyT.)
Contract number: 297347
Abstract: The presence of pharmaceuticals classified as emerging contaminants (EC) in surface water, groundwater, and drinking water generates uncertainty concerning the interactions that could be occurred with aquatic organisms and living beings. Thus, the monitoring of hospital wastewater is of great importance to identify the main classes of pharmaceuticals that could be discharged to the municipal sewage system and wastewater treatment plants (WWTPs). This work described the implementation and validation of a highly selective and sensitive analytical method using solid-phase extraction (SPE) and liquid chromatography-tandem mass spectrometry (LC-MS/MS) for the detection and quantification of these emerging compounds. The solid-phase extraction (SPE) method was employed using Oasis HLB cartridges. For LC-MS/MS analysis, the chromatographic separation was conducted in a C-18 Kinetex column (Phenomenex), and detection was achieved in an AB SCIEX QTrap 3200 tandem mass spectrometer (MS/MS) in the multiple reaction monitoring (MRM) mode. The quantitative analysis was performed by using the internal standard (IS) method with isotopically labeled analogs. The implemented method presented good linearity within the concentration range of 0.1-400 µg L-1 showing correlation coefficients (R2) ranged from 0.991 to 0.999. The limits of detection (LODs) were from 0.02 to 0.59 µg L-1, while the limits of quantification (LOQs) ranged from 0.07 to 1.80 µg L-1. The analytical method was successfully applied to the analysis of wastewater samples discharged by a public hospital in San Nicolas de los Garza, Nuevo Leon, Mexico, in two sampling periods: May 2017 and March 2018.
Keywords: Emerging contaminants, pharmaceuticals, hospital effluent, solid phase extraction, LC-MS/MS.
Resumen: La presencia de fármacos, clasificados como contaminantes emergentes, en agua superficial, subterránea y potable, genera incertidumbre sobre las interacciones que podrían ocurrir con organismos acuáticos y los seres vivos. Por lo tanto, el monitoreo del agua residual hospitalaria es de gran importancia para identificar los principales productos farmacéuticos que podrían descargarse al sistema de alcantarillado municipal y por lo tanto, estar presentes en el agua a tratar en las plantas de tratamiento de agua residual (PTAR). Este trabajo describe la implementación y validación de un método analítico altamente selectivo y sensible utilizando extracción en fase sólida (SPE) y cromatografía líquida acoplada a espectrometría de masas tándem (LC-MS/MS) para la detección y cuantificación de estos compuestos emergentes. Se empleó el método de extracción en fase sólida utilizando cartuchos Oasis HLB. Para el análisis LC-MS/MS, la separación cromatográfica se realizó en una columna Kinetex C-18 (Phenomenex), y la detección se realizó en un espectrómetro de masas en tándem AB SCIEX QTrap 3200 (MS/MS) en el modo de monitoreo de reacciones múltiples (MRM). El análisis cuantitativo se llevó a cabo utilizando el método de estándar interno (IS) con análogos marcados isotópicamente. El método presentó una buena linealidad dentro del rango de concentración de 0.1 a 400 µg L-1, con coeficientes de correlación (R2) que oscilaron entre 0.991 y 0.999. Los límites de detección (LOD) fueron de 0.02 a 0.59 µg L-1, mientras que los límites de cuantificación (LOQ) variaron de 0.07 a 1.80 µg L-1. El método analítico se aplicó con éxito al análisis de muestras de agua residual vertidas por un hospital público de San Nicolás de los Garza, Nuevo León, México, en dos períodos de muestreo: mayo de 2017 y marzo de 2018.
Palabras clave: Contaminantes emergentes, fármacos, efluentes hospitalarios, extracción en fase sólida, LC-MS/MS.
Introduction
The conservation of water resources is one of the main challenges in the world as a consequence of the use of water in all human activities, and due to their growing pollution, originated by the direct discharge of wastewater effluents in water bodies [1]. The recent monitoring of different water bodies and wastewater effluents has allowed the identification of organic contaminants in concentrations ranged from ng L-1-μg L-1, these compounds are known as emerging contaminants (EC). EC are substances that are not commonly monitored in the environment but have the potential to enter the environment and cause known or suspected adverse ecological and/or human health effects [2]. The introduction of the EC in water bodies generates concern respect to the toxicological effects in the aquatic organisms, due to the unlimited number of contaminants present in the discharges of wastewater effluents [3,4]. Some reports regarding the identification of EC in water bodies [5,6] and wastewater effluents [7,8] indicate that the EC with major occurrence are pharmaceuticals, drugs of abuse, pesticides, personal care products, flame retardants, industrial additives, and artificial sweeteners. Specifically, pharmaceuticals compounds were the first EC identified in the water surface, groundwater, and wastewater [9-11]; their introduction in the aquatic environment is mainly attributed to urban wastewater and wastewater treatment plants (WWTPs) effluents [12,13].
The variability in the number and concentrations of pharmaceuticals identified in the WWTPs effluents is a consequence of the mixture of different wastewater discharges, precisely, as domestic, industrial, and hospital effluents. Mainly, the water consumption in a hospital varies between 400-1200 L bed-1 day-1, generating a higher volume of wastewater with different therapeutic groups of drugs [14]. High concentrations of antibiotics, β-blockers, anti-inflammatory, and psychiatric drugs have been detected during determination of pharmaceuticals in hospital wastewater. In the last decade, the pharmaceuticals monitoring in hospital wastewater of various regions around the world (China, Switzerland, Portugal, Taiwan, USA, Spain, and Japan) confirms the presence of five main therapeutic groups of pharmaceuticals: antibiotics, anti-inflammatories, analgesics, antiepileptics and β-blockers [15-22].
In Mexico, the monitoring of pharmaceuticals in hospital wastewater is scarcely described. Few studies have been reported regarding pharmaceutical's determination in the wastewater of some hospitals in the central zone of the country [23-25]. The pharmaceuticals detected in these studies belong to anti-inflammatory, antidiabetic, β-blockers, and β-lactamic therapeutic groups. Therefore, in this work, a multi-class method was based on solid-phase extraction (SPE) followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The analytical method was applied to determine pharmaceuticals in wastewater discharge of a public hospital in San Nicolas de los Garza, Nuevo Leon, Mexico, in two different sampling periods: May 2017 and March 2018. The methodology was validated according to the guidelines established by the EPA, Method 1694: Pharmaceuticals and Personal Care Products in Water, Soil, Sediment and Biosolids by HPLC-MS/MS [26,27] which allows the detection and quantification of pharmaceuticals through a process of sample pre-treatment using SPE and determination by LC-MS/MS.
Experimental
Chemicals and reagents
The standard solutions were prepared using analytical reagents grade of high purity (>90%). Sulfamethoxazole, metformin, enalapril, metoclopramide, naproxen, caffeine, isoniazid, ampicillin, and loratadine were purchased from Sigma Aldrich. Omeprazole, ketoprofen, ibuprofen, acetaminophen, losartan, atenolol, and oxcarbazepine were purchased from USP Reference Standards. The stable isotope-labeled compounds carbamazepine-d2, levocetirizine-d4, acetaminophen-d4, and sulfamethoxazole-d4 (Toronto Research Chemical) were used as the internal standards. Individual standard solutions of all compounds were prepared in water at the concentration of 100 mg L-1. The working standard solutions were prepared at 1000 µg L-1 by mixing the appropriate amounts of the individual standard solutions and by diluting them with water/methanol (50:50, v/v). All solutions were preserved at 4 °C. All solvents were of LC-MS grade (>99%), manufactured by Sigma Aldrich, and the other reagents were of analytical grade. HPLC water was obtained by TEDIA (high purity solvents). The SPE extraction cartridges were Oasis HLB (N-vinylpyrrolidone-divinylbenzene phase balance) from Waters Corporation Milford, USA.
Sampling and wastewater characterization
The hospital is located in the urban area of San Nicolas de los Garza, Nuevo Leon, Mexico. It is a public hospital of the second level belonging to the Health Services of Nuevo Leon. It has 343 beds divided into nine departments. The wastewater samples were collected following the sampling described by Mexican guidelines (NMX-AA-003-1980) [28]. The samples were collected from the discharge effluent every hour (1 L h-1) for eight hours and stored in amber flasks at 4 °C for its transfer to the laboratory. The samples collected were mixed to obtain a composite sample. Once in the laboratory, the composite sample was filtered through a stainless steel micro-mesh to remove solids with large size, then it was filtered through 3 μm filter paper and filtered again, this time through a 0.45 μm nylon filter; the sample was stored at -20 oC.
The main physicochemical parameters were determined in the two composite samples. Biochemical oxygen demand (BOD) and chemical oxygen demand (COD) according the NMX-AA-028-SCFI-2001 and NMX-AA0-030-SCFI-2001 respectively. The inorganic ions (Cl-, SO 42-, NO3-, PO4 3- ) were determined by ion-exchange chromatography (Dionex ICS-110 system Thermo Fisher, USA). The chromatographic conditions for the inorganic ions analysis are described in Table 1 [29].
Chromatographic conditions for the determination of inorganic ions in the collected samples
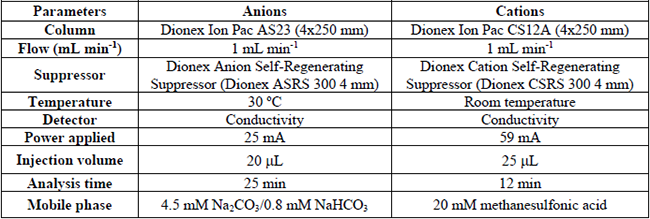
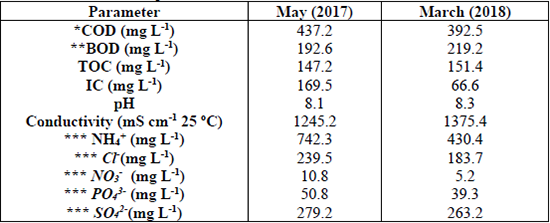
The conductivity and pH were determined by a portable meter Thermo Scientific Orion Star A329. The total organic carbon (TOC) and inorganic carbon (IC) were measured in a total organic carbon analyzer SHIMADZU VCSH. The main characteristics of the collected samples are shown in Table 2.
Results of wastewater samples characterization
Solid-phase extraction (SPE) procedure
For the drugs quantification, the SPE was carried out using the HLB cartridges (500 mg/ 6 cc) and the procedure was conducted according to the guidelines established by the EPA [26,27]. The HLB cartidges are the most widely used sorbents in SPE process [15-22]. The HLB polymer contains nonpolar (benzene and aliphatic chains) as well as polar (pyrrolidone) groups. Therefore, this sorbent has good ability for the interaction with both, polar and nonpolar organic molecules [30]. Initially, the Oasis HLB stationary phase was pre-conditioned by passing 4 mL of methanol and 6 mL of pure water; then, 200 mL of water sample (adjusted to pH 8 with ammonium hydroxide) was passed through the cartridge at a flow rate of 1 mL min-1. The stationary phase was dried under vacuum for 3 min, and the analytes were eluted using 5 mL of methanol at 1 mL min-1. The sample was injected into the chromatograph. The analysis was performed in triplicate. Sample recovery tests were done spiking the samples with sixteen pharmaceuticals and four isotopically labeled drugs at 200 µg L-1 level for each compound. The spiked samples were analyzed (n=3) by the proposed methodology.
LC-MS/MS conditions
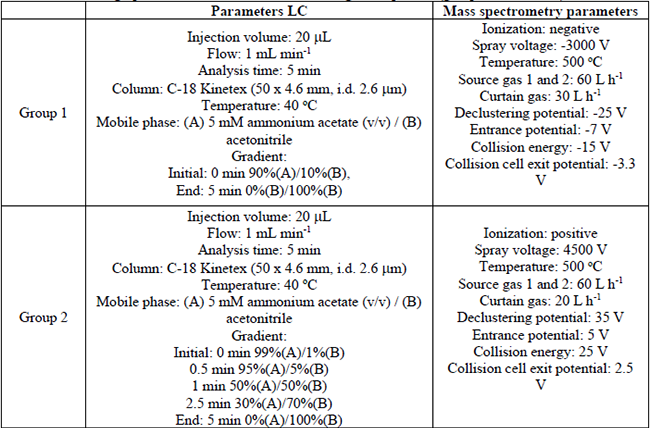
The chromatographic analysis was carried out by liquid chromatography-tandem mass spectrometry (LC-MS/MS) system operated in positive and negative ionization modes. Data acquisition was performed using Analyst software version 1.6. The chromatographic separation was performed in a Separations Module UFLC Shimadzu, an automatic injector, and a thermostatted column compartment (40 oC) and C-18 Kinetex (50 × 4.6 mm, i.d. 2.6 (m) column (Phenomenex). The mobile phase eluents were; (A) 5 mM ammonium acetate and (B) acetonitrile. During the chromatographic separation, the target compounds were subdivided into four groups (depending on mass spectrometry parameters). Depending on each group, the gradient elution program and analysis time were modified. The injection volume (20 µL) and flow rate (0.8 to 1.0 mL min-1) were similar for all the groups. The chromatography instrument was coupled to a mass spectrometer AB SCIEX QTRAP 3200 System triple quadrupole equipped with an electrospray ionization (ESI) source (Turbo IonSpray) and with continuous electron multiplier detector working in positive or negative ionization mode.
The triple quadrupole system allows the application of different operation MS/MS modes: product-ion scan, precursor-ion scan, neutral-loss scan and multiple reaction monitoring (MRM), which is the most predominant. Particularly, the MRM mode provides several advantages for quantification of organic pollutants, such as increased selectivity, reduced interferences, higher sensitivity and short analysis time [31]. Therefore, the pharmaceutical quantification was conducted in MRM mode using the protonated molecular ion as the precursor. The most abundant transition production was used for quantification (calibration curves), and the second transition was used for confirmation. Initially, the precursor ion (m/z) of the drugs was optimized in Q1 by direct infusion of individual dissolutions at 1000 µg L-1 for each compound in water/methanol (50:50, v/v). The parameters of the mass analyzer: spray voltage, temperature, curtain gas, source gas, entrance potential, collision cell exit potential, and collision energy were also optimized by direct infusion mode. In Q2, two transition fragments were selected and optimized for each analyte; the operation mode was product ion. After the optimization of the collision energy in Q2, two different transitions (precursor ion-product ion) were selected for each compound, one for quantification (transition 1) and one for confirmation (transition 2), and those ions were monitored in time-scheduled MRM conditions.
Method validation
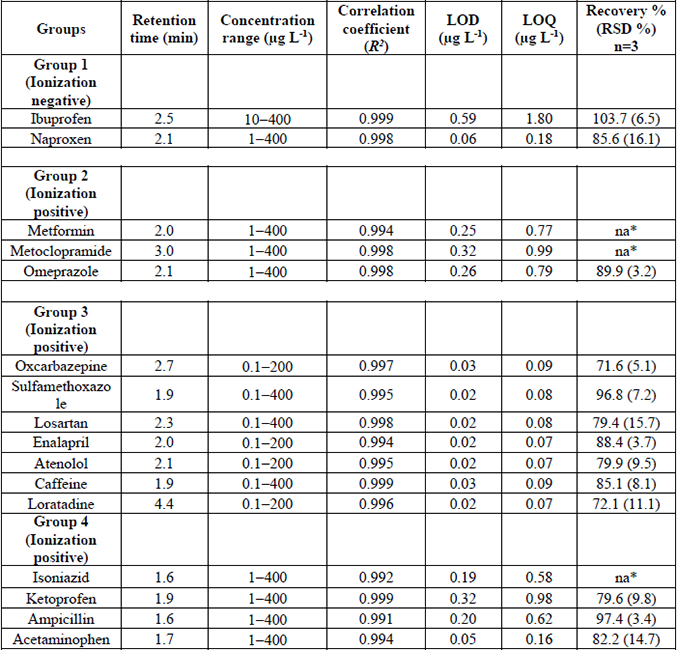
The analytical method was validated through various parameters such as linearity, the limits of detection (LODs), the limits of quantification (LOQs), extraction recovery, and signal suppression. For LC-MS/MS quantification, the internal standard (IS) method was used to compensate for the matrix effect during the determination of pharmaceuticals in wastewater. The correction of the matrix effects was carried out with isotopically labeled compounds, one for each group of drugs (levocetirizine-d4 group 1, carbamazepine-d2 group 2, sulfamethoxazole-d4 group 3, and acetaminophen-d4 group 4). The selection criterion was based depending on the similitude in the mass spectrometer parameters and chemical structure for each group of drugs and the labeled compound.
For the IS calibration, standard solutions of each compound were prepared in pure water; the corresponding IS was spiked at 200 µg L-1 in all the concentration levels of the calibration standards for each compound. The ratio of the compound area / IS area at each concentration level was considered to generate the calibration curves. Linearity was evaluated according to the correlation coefficient (R2) for linear regression by injecting the standard solutions of each analyte in triplicate at five concentration levels. The concentration range was from 0.1 to 400 µg L-1. The LODs and LOQs were estimated by the injection of standard solutions at the lowest concentration level corresponding to a signal-to-noise ratio > 3 for LOD and > 10 for LOQ, respectively [17,27]. The precision was expressed as repeatability in terms of relative standard deviation (RSD). Recovery percentages in hospital wastewater samples spiked with target compounds at a concentration level of 200 µg L-1 each compound was used to evaluate the accuracy of the method. The samples were analyzed in triplicate by LC-MS/MS before and after spiking process. The extraction recoveries were calculated as the ratio between the compound concentration after SPE processes (CA, spiked sample) subtracted by the concentration detected in the hospital samples (CB, non-spiked sample) and the theoretical concentration added in hospital wastewater (CC) (Eq. 1) [17].
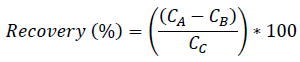 [Eq. 1]
[Eq. 1]According to the IUPAC criteria, the mean recovery of the replicate values must be within ± 30% of the theoretical concentration added [32]. According to the Eq. 2, the ion suppression (expressed as a percentage) was evaluated: the peak area in hospital wastewater extracts spiked with target compounds (AA) are first subtracted by the peak area of the compound in the non-spiked hospital wastewater extracts (AB), the values obtained are then compared with the peak area of the analyte in the standard solution water/methanol (50:50, v/v) spiked with the target compounds (AC) [33].
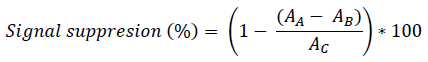 [Eq. 2]
[Eq. 2]Results and discussion
LC-MS/MS analysis
The analytical method using SPE and LC-MS/MS was implemented to detect pharmaceuticals compounds contained in a discharge of a public hospital in Mexico. The target compounds were selected mainly based on their detection and occurrence in hospital effluents, urban wastewater, and WWTP effluents in Mexico and around the world. Initially, thirty pharmaceutical compounds of fifteen different therapeutic groups were selected, and a screening analysis was conducted through LC-MS/MS system. The information of precursor ion and two different transitions for each compound were collected for the monitored pharmaceutical compounds. This information was processed in LC-MS/MS system and analyzed through of MRM mode operation. Thus, the capability of the instrument to detect sixteen compounds of different therapeutic groups: sulfamethoxazole, metformin, enalapril, metoclopramide, naproxen, caffeine, isoniazid, ampicillin, loratadine, omeprazole, ketoprofen, ibuprofen, acetaminophen, losartan, atenolol, and oxcarbazepine were demonstrated. These compounds were selected for the method implementation. Once selected, for the MS/MS settings, the specific precursor ion, and two transition fragments were optimized for the twenty pharmaceuticals (sixteen compounds and four internal standards), as is shown in Fig. 1 for naproxen, as an example.
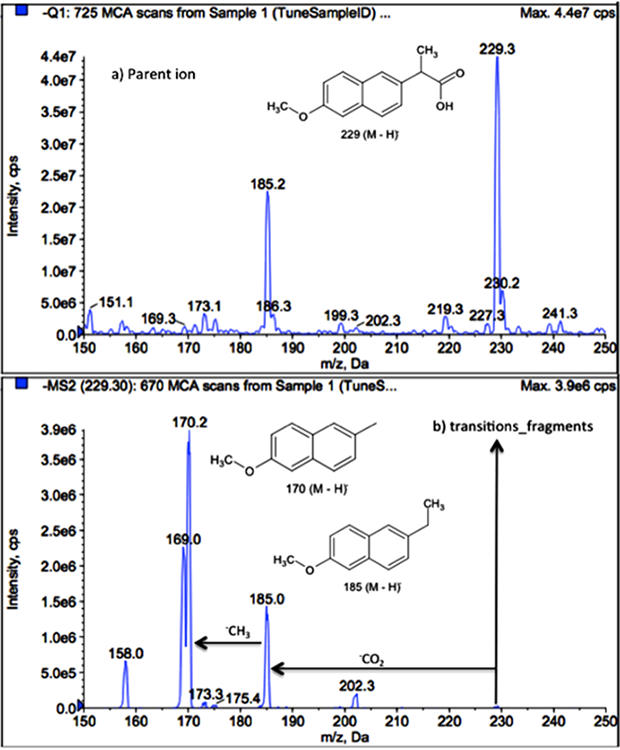
Fig. 1
Example of signal optimization for naproxen during direct infusion mode: (a) MS spectrum and (b) MS/MS spectrum for naproxen.
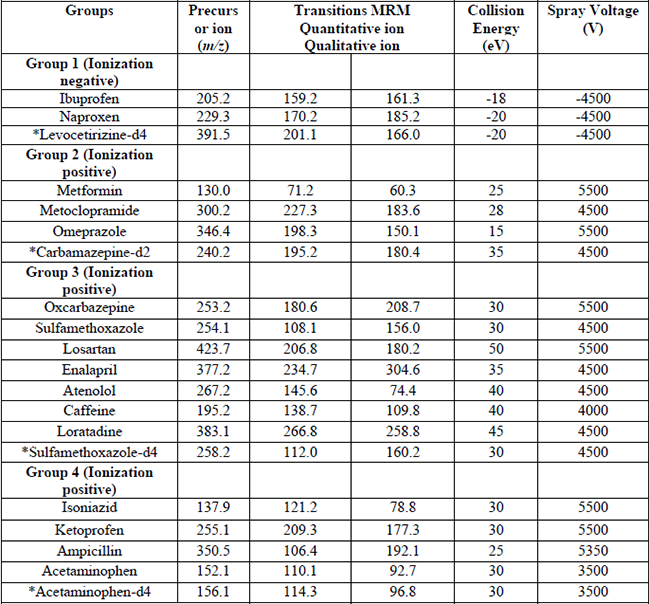
The LC-MS/MS parameters for the pharmaceuticals were conducted under ESI. Initially, the analytical signal was evaluated in ESI positive and negative ion modes. The selected precursor ions for all the compounds showed maximum sensitivity in positive ionization mode. Only, the naproxen, ibuprofen, and levocetirizine-d4 were optimized in ionization negative. Subsequently, precursor ion, transition fragments (MRM), ionization mode, and conditions of collision energy (eV) and spray voltage were obtained for each compound; this information was processed in the LC-MS/MS system for the detection of each pharmaceutical in MRM mode. On the other hand, the analytes were subdivided into four groups for chromatographic separation, considered their LC-MS/MS parameters (ionization mode, collision energy, spray voltage) and guidelines established by the EPA (Method 1694). A summary of the mass spectrometric parameters including the precursor ion, the transition fragments (MRM), the ionization mode, the collision energy (eV), spray voltage, and pharmaceuticals groups are shown in Table 3.
Specific MS/MS settings for the sixteen selected pharmaceuticals compounds
For chromatographic analysis, the elution of the compounds was conducted in four groups using reversed-phase C-18 analytical column and mobile phases based on 5 mM ammonium acetate (A)/acetonitrile (B) mixture (Table 4).
Chromatographic conditions for the elution of target compounds (groups 1, 2 ,3 and 4).
Chromatographic conditions for the elution of target compounds (groups 1, 2 ,3 and 4). (Cont.)
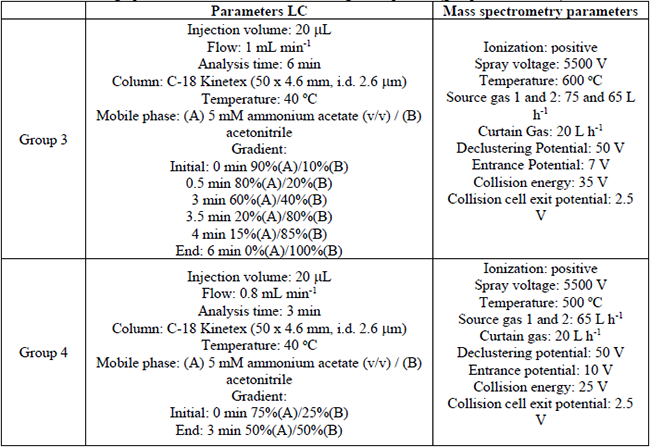
Several linear gradients were tested to obtain the best chromatographic separation. The mass spectrometry conditions were similar for the four pharmaceutical groups; the ionization mode was negative (group 1) and positive (group 2, 3, and 4). The spray voltages were: -3000 V (group 1), 4500 V (group 2) and 5500 V (group 3 and 4). The collision energy for group 1 was -15 V; 25 V for group 2 and 4; and 35 V for group 3. In general, the same conditions LC-MS/MS analysis were established for the four groups. In Figures 2 and 3 the MRM chromatograms of the four groups are depicted.
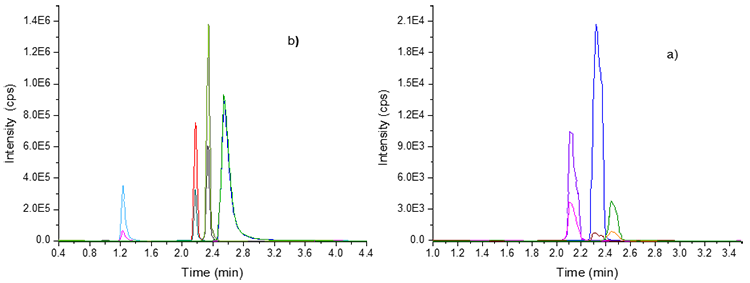
Fig. 2
MRM extracted chromatogram of target analytes of: (a) Group 1; naproxen (tR=2.1 min), *levocetirizine-d4 (2.3 min), ibuprofen (tR=2.45min) (b) Group 2; metformin (tR=1.3 min), omeprazole (tR=2.2 min), *carbamazepine-d2 (tR=2.3 min), metoclopramide (tR=2.6 min)
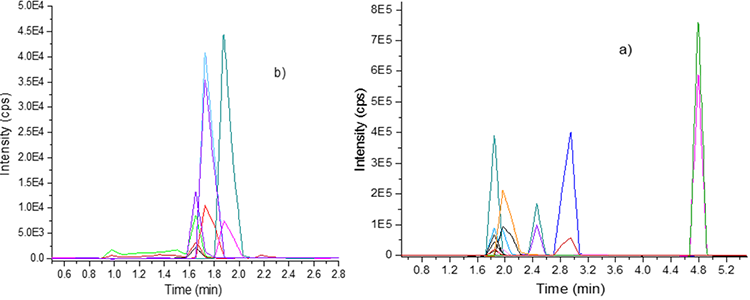
Fig. 3
MRM extracted chromatograms of target analytes of: (a) Group 3; caffeine (tR=1.9 min), sulfamethoxazole (tR=1.9 min), *sulfamethoxazole-d4 (tR=1.9 min), enalapril (tR=2.0 min), atenolol (tR=2.0 min), losartan (tR=2.5 min), oxcarbazepine (tR=3.0 min), loratadine (tR=4.8 min); (b) Group 4; ampicillin (tR=1.65 min), isoniazid (tR=1.65 min), acetaminophen (tR=1.7 min), *acetaminophen-d4 (tR=1.7 min), ketoprofen (tR=1.9 min).
For the four groups, the elution of target compounds was conducted in linear-gradient, the flow rates ranging from 0.8 to 1 mL min-1, and retention times (tR) ranging from 1 to 5 min. The MRM analysis allowed the detection of two transitions for each pharmaceutical. Therefore, during the elution were observed two characteristics signals for each compound: quantification (most abundant) and confirmation (less abundant). Despite the chromatographic gradient used, coelution was encountered (groups 3 and 4). However, these compounds could be easily identified by their precursor ion (accurate masses) and two transitions (MRM). Specifically, the elution of the compounds of group 1: naproxen, ibuprofen, and levocetirizine-d4 (Figure 2(a)) were conducted in tR ranging from 2.1 to 2.4 min, while that the analytes of group 2: metformin, omeprazole, *carbamazepine-d2 and metoclopramide (Figure 2(b)) were eluted in tR ranging from 1.3 to 2.6 min. On the other hand, the elution of the compounds of group 3: caffeine, sulfamethoxazole, *sulfamethoxazole-d4, enalapril, atenolol, losartan, oxcarbazepine, and loratadine was conducted in tR ranging from 1.9 to 4.8 min (Figure 3(a) ). In contrast, the analytes of group 4: ampicillin, isoniazid, acetaminophen, *acetaminophen-d4, and ketoprofen (Figure 3(b) ) were eluted in tR ranging between 1.6 and 1.9 min. Therefore, the target compounds were separated under the proposed chromatographic conditions. The short analysis time (< 6 min) per group, use of simple elution gradients, detection of different therapeutic groups of pharmaceuticals, and high speed of analysis were some advantages of the implemented analytical method.
Method Validation
Matrix effects such as signal suppression are often the concern of LC-MS/MS analysis during the detection of organic pollutants. The hospital wastewater is a mixture of other different pollutants. Therefore, during the analysis of pharmaceuticals by LC-MS/MS, these compounds mean matrix effects and interferences that are present in the sample. Additionally, they affect ionization (ESI) and the detection of organic compounds [34]. Therefore, ion suppression owing to matrix effects was calculated with the Eq. 2. A summary of the ion suppression percentage of analyzed compounds is shown in Fig. 4.
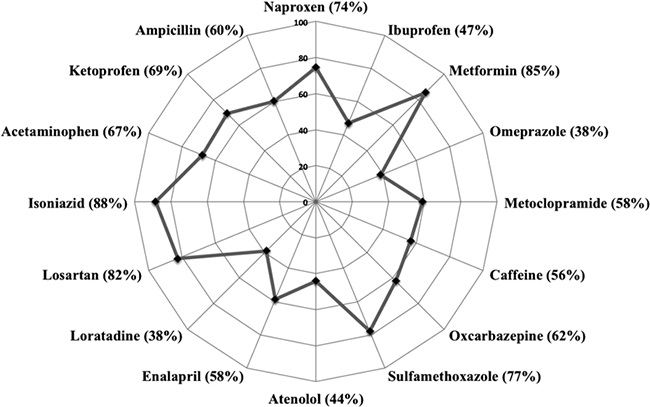
Fig. 4
Matrix effects (ion suppression) of the target compounds in hospital wastewater.
The ions suppression ranged from 38 to 88% (Fig. 4). Losartan, isoniazid, and metformin showed the highest suppression values (82-88%). These results indicate that the presence of different types of pollutants (disinfectants, metals, detergents, organic and inorganic ions) in hospital wastewater contributes to the loss of sensitivity and accuracy during the detection and quantification of pharmaceuticals by LC-MS/MS [14,17]. Therefore, the implementation of the IS method with isotopically labeled analogs for quantification by LC-MS/MS was a powerful tool to compensate adverse matrix effect in hospital wastewater samples. Subsequently, the concentrations of the detected drugs were calculated using a standard calibration curve using IS method. Only the most abundant transition (quantification) was employed. The calibration curves for the four groups ranged from 0.1 to 400 µg L-1, depending on the sensitivity of each compound.
The calibration curves were well adjusted to a linear model with correlation coefficients (R2) between 0.991 to 0.999 for the analyzed compounds. The LODs and LOQs ranged from 0.02 to 0.59 µg L-1 and 0.07 to 1.80 µg L-1, respectively. The overall precision of the method, expressed as the relative standard deviation (RSD) of repeatability at 1.0 µg L-1 fortification level was satisfactory, ranging from 0.9 to 12.7%. On the other hand, accuracy of the method was estimated from recovery experiments: acceptable recovery values of the spiked samples of hospital wastewater with the target compounds were obtained within the range of 72 to 103%, exhibiting RSD values ranging from 3.2% (omeprazole) to 14.7% (acetaminophen). Only, naproxen (16.1%) and losartan (15.7%) obtaining values above 15%. Unfortunately, the recovery values for isoniazid, metoclopramide, and metformin were unacceptable (< 45%); therefore, these compounds were not quantified in the wastewater samples. The analytical parameters for analyzed pharmaceuticals are summarized in Table 5.
Validation parameters of the implemented LC-MS/MS method for the pharmaceuticals determination in hospital wastewater.
Quantitative analysis of detected drugs
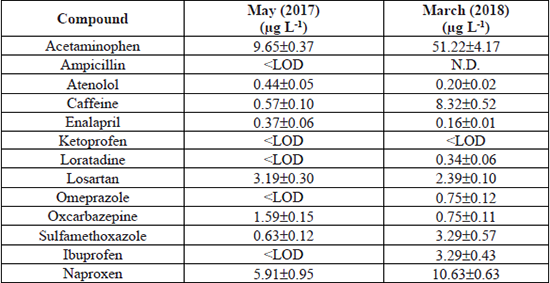
The proposed analytical method was applied to quantify sixteen pharmaceuticals in a wastewater discharge of a public hospital in San Nicolas de los Garza, Nuevo Leon, Mexico, in two different sampling periods: May 2017 and March 2018. From the analyzed compounds, sixteen were detected in samples of May 2017, while in samples of March 2018 were identified fifteen compounds. Although isoniazid, metformin, and metoclopramide were detected in both samples; as was stated before, their recovery values were low (< 45 %); therefore, they were not considered for quantification analysis. Specifically, eight pharmaceuticals (acetaminophen, atenolol, caffeine, enalapril, losartan, oxcarbazepine, sulfamethoxazole, and naproxen) were quantified in concentrations ranging from 0.37- 9.65 µg L-1 in the samples collected in May 2017. The other five compounds (ampicillin, ketoprofen, loratadine, omeprazole, and ibuprofen) were below of LOD (< 0.59 µg L-1).
The eight drugs detected in the previous period coincide with those identified in the hospital effluent of March 2018; however, loratadine, omeprazole, and ibuprofen were additionally detected. The concentration ranges of the eleven compounds quantified were from 0.16-51.2 µg L-1. The drug ketoprofen was below of LOD (< 0.32 µg L-1), while the ampicillin was not detected in the second sampling period. Acetaminophen showed the highest concentration in the samples collected in May 2017 and March 2018: 9.6 µg L-1 and 51.2 µg L-1, respectively. A summary of the quantified concentrations in the two collected samples is shown in Table 6.
Concentration of the determined pharmaceuticals in the hospital wastewater
In Mexico, the monitoring of pharmaceuticals in hospital wastewater is scarce. Only few studies have been reported for the determination of pharmaceuticals in wastewater from some hospitals in two regions of the central zone of the country: State of Mexico and Puebla [23-25,35]. In these studies, only five drugs (acetaminophen, atenolol, ibuprofen, naproxen and omeprazole) were detected in concentrations ranging from 0.009 to 123 µg L-1 (Table 7).
Concentrations of the detected pharmaceuticals in hospital wastewater in other regions of Mexico (µg L-1).
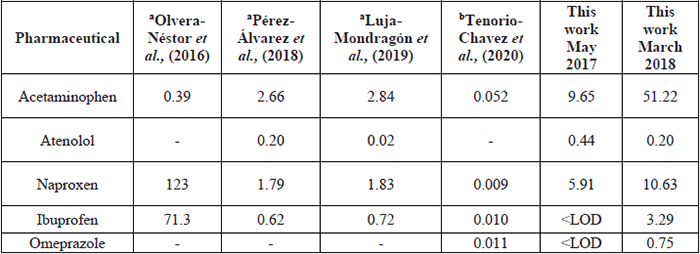
Naproxen was reported at the highest concentration (123 µg L-1) in those studies, and the value was ten-twenty times higher than values described in this work (5.91 µg L-1 and 10.63 µg L-1). However, acetaminophen was quantified in lower concentrations (0.052-2.84 µg L-1) than those detected in this research (9.65 µg L-1 and 51.22 µg L-1). Therefore, the present study confirms the variability and concentration range of detected drugs in hospital wastewater, demonstrating the necessity of in-depth research in Mexico of pharmaceutical monitoring in these wastewater samples. These compounds are directly discharged in the municipal sewage system and, subsequently, seen in the WWTP system, where its removal is incomplete.
Conclusion
The monitoring of pharmaceuticals in wastewater from a public hospital in San Nicolas de los Garza, Nuevo Leon, Mexico was conducted in this study. The implementation of a selective and sensitive analytical method through the SPE process and LC-MS/MS system allows the detection of drugs belonging to different therapeutic groups. In the samples collected, sixteen compounds were detected in concentrations ranging from 0.16-51.2 µg L-1. The higher level of pharmaceuticals was detected in March 2018, which can be directly associated with the high number of patients in the hospital in this period. Additionally, the acetaminophen drug showed the highest concentration in both sampling periods.
Acknowledgments
The authors thank the financial support from Programa de Apoyo a la Investigación Científica y Tecnológica PAICYT-UANL. R. Hernández-Tenorio acknowledges his doctoral scholarship (grant number 297347) from Consejo Nacional de Ciencia y Tecnología (CONACyT.) The authors would like to thank the public hospital to provide the facilities for wastewater sampling.
References
1. Tran, N.H.; Reinhard M.; Gin K.Y.H., Water Res. 2018, 133, 182-207. DOI: https://doi.org/10.1016/j.watres.2017.12.029
2. Verlicchi, P. Hospital Wastewaters. Ed. Springer, 2018.
3. Gavrilescu, M.; Demnerová, K.; Aamand, J.; Agathos, S.; Fava, F., New Biotechnol. 2015, 32, 147-156. DOI: https://doi.org/10.1016/j.nbt.2014.01.001
4. Richardson, S.D.; Kimura, S.Y. Environmental Technology & Innovation 2017, 8, 40-56. DOI: 10.1016/j.eti.2017.04.002
5. Lapworth, D.J.; Baran, N.; Stuart, M.E.; Manamsa, K.; Talbot, J. Environ. Pollut. 2015, 203, 214-225. DOI:10.1016/j.envpol.2015.02.030
6. Meador, J.P.; Yeh, A.; Young, G.; Gallagher, E.P. Environ. Pollut. 2016, 213, 254-267. DOI: 10.1016/j.envpol.2016.01.088
7. Loos, R.; Carvalho, R.; António, D.; Comero, S.; Locoro, G.; Tavazzi, S.; Paracchini ,B.; Ghiani, M.; Lettieri,T.; Blaha, L.; Jarosova, B.; Voorspoels, S.; Servaes ,K.; Haglund, P; Fick, J.; Lindberg, R.; Schwesing, D.; Gawlik, B. Water Res. 2013, 47, 6475-6487. DOI: 10.1016/j.watres.2013.08.024
8. Archer, E.; Petrie, B.; Kasprzyk-Hordern, B.; Wolfaardt, G.M. Chemosphere 2017 174, 437-446. DOI: 10.1016/j.chemosphere.2017.01.101
9. Halling-Sørensen, B.; Nielsen, S.N.; Lanzky, P.F.; Ingerslev, F.; Lützhøft, H.H; Jørgensen, S, Chemosphere 1998, 36, 357-393. DOI: 10.1016/s0045-6535(97)00354-8
10. Ternes, T.A. Water Res. 1998, 32, 3245-3260. DOI: https://doi.org/10.1016/S0043-1354(98)00099-2
11. Kummerer, K.; Erbe, T.; Gartiser, S.; Brinker, L. Chemosphere 1998, 36, 2437-2445. DOI: 10.1016/s0045-6535(97)10200-4.
12. Sui,Q.; Cao, X.; Lu, S.; Zhao, W.; Qiu, Z.; Yu, G. Emerging Contaminants 2015, 1, 14-24. DOI: https://doi.org/10.1016/j.emcon.2015.07.001
13. Sangion, A.; Gramatica, P. Environ. Res. 2016, 147, 297-306. DOI: 10.1016/j.envres.2016.02.021
14. Perrodin, Y.; Christine, B.; Sylvie, B.; Alain, D; Jean-Luc, B.K.; Cécile, C.O.; Elodie, B. Chemosphere 2013, 90, 1037-1046. DOI: https://doi.org/10.1016/j.chemosphere.2012.08.049
15. Yuan, S.; Jiang, X.; Xia, X.; Zhang, H.; Zheng, S. Chemosphere 2013, 90, 2520-2525. DOI: https://doi.org/10.1016/j.chemosphere.2012.10.089
16. Kovalova, L.; Siegrist, H.; Von Gunten, U.; Eugster, J.; Hagenbuch, M.; Wittmer, A.; McArdell, C.S. Sci.Total Environ. 2013, 47,7899-7908. DOI: 10.1021/es400708w
17. Gros, M.; Rodríguez-Mozaz, S.; Barceló, D. J. Chromatogr. A 2013, 1292, 173-188. DOI: 10.1016/j.chroma.2012.12.072
18. Santos, L.H.; Gros, M.; Rodriguez-Mozaz, S.; Delerue-Matos, C.; Pena, A.; Barceló, D.; Montenegro, M.C.B. Sci. Total Environ. 2013, 461, 302-316. DOI: 10.1016/j.scitotenv.2013.04.077
19. Li, SW.; Lin, A.Y.C Chemosphere 2015, 139, 190-196. DOI: https://doi.org/10.1016/j.chemosphere.2015.06.010
20. Oliveira, T.S.; Murphy, M.; Mendola, N.; Wong, V.; Carlson, D.; Waring, L. Sci Total Environ 2015, 518,459-478. DOI: 10.1016/j.scitotenv.2015.02.104
21. Mendoza, A.; Aceña, J.; Pérez, S.; de Alda, M.L.; Barceló, D.; Gil, A.; Valcárcel, Y. Environ. Res. 2015, 140, 225-241. DOI: 10.1016/j.envres.2015.04.003
22. Azuma, T.; Arima, N.; Tsukada, A.; Hirami, S.; Matsuoka, R.; Moriwake, R.; Mino, Y. Sci. Total Environ. 2016, 548, 189-197. DOI: 10.1016/j.scitotenv.2015.12.157
23. Olvera-Néstor, C.G.; Morales-Avila, E.; Gómez-Olivan, L.M.; Galár-Martínez, M.; García-Medina, S.; Neri-Cruz, N. Bull. Environ. Contam. Toxicol. 2016, 96, 326-332. DOI: https://doi.org/10.1007/s00128-015-1721-3
24. Pérez-Alvarez, I.; Islas-Flores, H.; Gómez-Oliván, L.M.; Barceló, D.; De Alda, M.L.; Solsona, S.P.; Galar-Martínez, M. Environ. Pollut. 2019, 240, 330-341. DOI: 10.1016/j.envpol.2018.04.116
25. Luja-Mondragón, M.; Gómez-Oliván, L.M.; SanJuan-Reyes, N.; Islas-Flores, H.; Orozco-Hernández, J.M.; Heredia-García, G.; Dublán-García, O. Sci. Total Environ. 2019, 660, 751-764. DOI: 10.1016/j.scitotenv.2019.01.072
26. Englert, B. US Environ. Prot. Agency Technology OOSA 1-72, 2007.
27. Ferrer, I.; Zweigenbaum, J.A.; Thurman, E.M. J. Chromatogr. A , 2010, 1217, 5674-5686. DOI: 10.1016/j.chroma.2010.07.002
28. NMX-AA-030-SCFI-2001. Norma Oficial Mexicana. Determinación de la demanda química de oxígeno en aguas naturales, residuales y residuales tratadas. Procuraduría Federal de Protección al Ambiente. Diario Oficial de la Federación.
29. Access https://assets.thermofisher.com/TFS-Assets/CMD/Application-Notes/AN-LPN-3085-Beverages-Applications-Notebooks-Carbonated-LPN3085.pdf, accesed in October 2020
30. Jeong, Y.; Schaffer, A.; Smith, K. Chemosphere 2017, 174, 297-305. DOI: 10.1016/j.chemosphere.2017.01.116
31. Bletsou, A.A.; Jeon, J.; Hollender, J.; Archontaki, E.; Thomaidis, N.S. TrAC Trends in Anal. Chem. 2015 ,66, 32-44. DOI: https://doi.org/10.1016/j.trac.2014.11.009
32. Thompson, M.; Ellison, S.L.; Fajgelj, A.; Willetts, P.; Wood, R. Pure Appl. Chem. 1999, 71, 337-348. DOI: http://dx.doi.org/10.1351/pac199971020337
33. Kafeenah, H.I.; Osman, R.; Bakar, N.K.A. RSC advances 2018, 8, 40358-40368. DOI: 10.1039/C8RA06885B
34. Nödler, K.; Licha, T.; Bester, K.; Sauter, M. J. Chromatogr. A 2010, 1217, 6511-6521. DOI: 10.1016/j.chroma.2010.08.048
35. Tenorio-Chávez, P.; Cerro-López, M.; Castro-Pastrana, L.I.; Ramírez-Rodrigues, M.M.; Orozco-Hernández, J.M.; Gómez-Oliván, L.M. Sci. Total Environ. 2020, 727, 138716. DOI: 10.1016/j.scitotenv.2020.138716
Author notes
*Corresponding author: Aracely Hernández-Ramírez, email: [email protected] , Phone: 8183294010 ext. 3438
