Journal of the Mexican Chemical Society, vol. 65, no. 1, 2021
Sociedad Química de México A.C.
Daniel G. Araiza *
Universidad Nacional Autónoma de México
Francisco González-Vigi
Universidad Nacional Autónoma de México
Antonio Gómez-Cortés
Universidad Nacional Autónoma de México
Jesús Arenas-Alatorre
Universidad Nacional Autónoma de México
Gabriela Díaz *
Universidad Nacional Autónoma de México
Received: 15 July 2020
Accepted: 24 September 2020
Funding
Funding source: CONACyT
Contract number: 308329
Abstract: Platinum catalysts (1.5 wt. %) supported over CeO2 and γ-Al2O3 were synthesized via wet impregnation using two different Pt precursors: H2PtCl6 and Pt(acac)2. Catalysts were tested in the dry reforming of methane (DRM) reaction at stoichiometric conditions (CH4/CO2 molar ratio of 1) with two approaches: as a function of temperature (400-800 °C) and as a function of time on-stream (800 °C / 24 h). Platinum supported over ceria catalysts showed better catalytic properties, especially in the stability tests, where deactivation was almost negligible. In contrast, alumina-supported catalysts stability was considerably lower due to the increased formation of carbon residues and the significant Pt particle sintering after reaction at 800 °C for 24 h. Through different characterization techniques (TEM, CO chemisorption), a strong Pt-Ceria interaction was evidenced, which helped in preventing Pt particle agglomeration under reaction conditions and promoted active interface sites. Both features are proposed to be responsible for the Pt/CeO2 catalysts better catalytic performance. The effect of the Pt precursor depends on the nature of the support. In ceria, Cl species benefited the generation of oxygen vacancies and surface ceria reducibility; both features are responsible for the Pt/CeO2 anti-coke properties, thus impacting positively in the catalytic performance of the Pt(-cl)/Ce sample. Conversely, in alumina, these Cl species triggered particle sintering and carbon deposition during the DRM reaction, affecting the Pt(-cl)/Al catalytic performance.
Keywords: Methane dry reforming, Pt/Ceria, metal-support interaction, catalytic stability.
Resumen: Catalizadores de platino (1.5 % en peso) soportados sobre CeO2 y γ-Al2O3 fueron sintetizados mediante impregnación húmeda utilizando dos diferentes precursores de Pt: H2PtCl6 and Pt(acac)2. Los catalizadores fueron evaluados en la reacción de reformado seco de metano (DRM) en condiciones estequiométricas (razón molar de CH4/CO2 igual a 1) y con dos metodologías: en función de la temperatura de reacción (400-800 °C) y en función del tiempo de reacción (800 °C / 24 h). Los catalizadores de platino soportados sobre ceria mostraron las mejores propiedades catalíticas, especialmente en las pruebas de estabilidad, donde la desactivación fue muy baja. Por el contrario, la estabilidad catalítica de las muestras soportadas en alúmina fue considerablemente menor, debido tanto a la formación de residuos de carbón como al sinterizado de partículas de Pt. Por medio diferentes técnicas de caracterización (TEM, Quimisorción de CO), se evidenció una fuerte interacción Pt-Ceria, la cual ayudó a prevenir la aglomeración de partículas de Pt durante la reacción, además de promover la generación de sitios activos interfaciales. Ambas características se proponen como las responsables de las mejores propiedades catalíticas presentadas por los catalizadores Pt/CeO2. El efecto del precursor del Pt depende de la naturaleza del soporte. En ceria, las especies de cloro beneficiaron la generación de sitios vacantes de oxígeno así como la reducción superficial de la ceria; ambas características son responsables de las propiedades anti-coque en el sistema Pt/CeO2, por lo tanto, estas impactaron positivamente en el desempeño catalítico de la muestra Pt(-cl)/Ce. Por el contrario, en la alúmina, estas especies cloradas aparentemente promovieron el sinterizado de partículas y los depósitos de carbono durante la reacción, lo cual afectó el desempeño catalítico de la muestra Pt(-cl)/Al.
Palabras clave: Reformado seco de metano, Pt/Ceria, interacción metal-soporte, estabilidad catalítica.
Introduction
The concentration of greenhouse gases (GHG) in the atmosphere has increased importance in the last decades, mostly from anthropogenic activities [1]. GHG are responsible for global warming and climate-change issues; thus, strategies for limiting their release and diminishing their concentration in the environment are pivotal to solving their negative impacts [2]. A catalytic reaction suitable for the mitigation of the most abundant GHG, carbon dioxide and methane, is the dry reforming of methane (DRM), Eq. (1) [3]. Although the DRM is an endothermic reaction, it is considered a sustainable process since it reduces pollutants from the atmosphere (CH4 and CO2) while producing synthesis gas (syngas), a mixture of H2 and CO known as a building block to produce value-added compounds like alcohols and olefins through the Fischer-Tropsch process.[4-6]
 (1)
(1)Due to the highly endothermic nature of the reaction, it requires elevated temperatures (> 700 °C) to reach substantial conversion levels [7]. Under these hard-working conditions, catalysts experience some drawbacks, like coke deposition or metal particles sintering. The former comes from side reactions like the CH4 decomposition and the CO disproportionation [8], while the latter occurs in most of the transitions metals (including noble metals) when they are submitted to high temperatures [9]. The sintering of the metal active phase promotes the agglomeration and growth of the particles and diminishes the active sites where the reactants can adsorb and react. On the other hand, the formation of carbon residues during the DRM reaction blocks the active catalyst sites, affecting their catalytic performance. These two phenomena negatively impact the catalytic properties of materials.
Thus, the employed catalysts should be resistant to these highly demanding operations. Different types of supported metal catalysts such as Pt, Rh, Ir, Ni and Co-based have displayed promising catalytic properties in the DRM process [10]. Specifically, noble metals like platinum exhibit higher catalytic activity and stability due, among other things, to their higher resistance to carbon deposition [11]. On the other hand, although supported nickel catalysts have been extensively used in the DRM because of their low cost and reasonable activity [12], they are prone to deactivate very fast mainly to the deposition of carbon residues, which depends on the particle size of Ni [13]. Although platinum is considered one of the best choices for supported catalysts in the DRM reaction [14,15], it has been reported that Pt nanoparticles begin to sinter and agglomerate when heated to considerable high temperatures (> 500 °C) [16,17]. The above indicates that deactivation problems could be still encountered in the Pt-based catalysts, in agreement with our previous work [18].
Three factors are crucial to achieving competitive performance in the DRM reaction: i) the high dispersion of the metal, ii) the selection of adequate support, and iii) the degree of interaction between the metal and the support. The first feature impacts the catalyst anti-coke properties since high metal dispersion helps to inhibit the carbon formation during the DRM reaction [19]. Regarding the second, it is well-known that the nature of the support not only influences the metal dispersion and stabilization of the active phase but also participates in the reaction [20,21]. Finally, the third feature leads to the development of a metal-support interaction, positively impacting the catalytic performance, while inhibiting the sintering of the active phase [22]. These metal-support interactions can be developed on mildly acidic (Al2O3) or basic (CeO2) supports and become an important factor in the catalytic properties [23,24].
The metal-support interface regulates the catalytic properties by several ways, for example modifying electronic and geometric parameters; the former characterized by charge transfer between the metal sites and the support, while the latter, by decoration or coverage of metal sites by the support [25]. The classical strong metal-support interaction (SMSI) identified by Tauser and co-workers [26], involved noble metals supported on a reducible substrate (the representative example is TiO2) [27,28]; the partial reduction of the support at high temperatures severely affected the chemisorptive properties of the metal. Subsequently, the definition of this strong metal-support interaction was extended to include interactions between the metallic nanoparticles and any support exhibiting similar phenomena [29,30].
Typically, catalysts are prepared by depositing the metal particles onto high-surface-area supports to increase its dispersion. The use of γ-Al2O3 as a support promotes the above since it typically exposes a high surface area [31]. Nevertheless, its acidic nature triggers the formation of carbonaceous species in reforming reactions, and consequently, catalyst deactivation. The Pt/Al2O3 system has been studied in the DRM reaction with good performances due to the high dispersion of platinum [32,33]. A variety of platinum precursors have been used, exhibiting different properties; for example, chloride precursors promote the Pt dispersion but enhance the alumina acidity [32].
On the other hand, due to the enhanced oxygen storage capacity (OSC) of CeO2, coming from the redox Ce4+/Ce3+ couple [34], its use as a support has been beneficial in the control or removal of carbon deposits in reforming reactions, improving the stability of catalysts [18,35]. However, the preparation of high-surface ceria is challenging [36], and the material typically exhibits a low specific area, which could impact negatively the dispersion of the metal. Despite the above, high dispersion of noble-metal catalysts has been observed in ceria-supported materials [18]. The Pt-ceria system exhibits high activity and stability in the DRM reaction, mainly when a strong metal-support interaction is generated [14,37]. Nagai and co-workers [38] observed that the Pt-O-Ce bond acted as an anchor and inhibited Pt particles sintering in CeO2; these results show the importance of metal-support interaction related to the performance of the catalyst.
The preparation of supported metal catalysts is achieved through various methods; among them, the impregnation route has been widely employed mostly due to the easiness in obtaining well-dispersed metallic particles [39]. Traditionally, water-soluble metal precursors such as chlorinated compounds (e.g., H2PtCl6) are used, among others. In the search to improve the catalytic properties of materials, metallic organometallic precursors have also been explored, such as acetylacetonate (e.g., Pt(acac)2). Their main characteristic is that they do not contain chlorine, although they are only soluble in organic solvents.
In this frame, our aim was to evaluate the nature of the support (alumina or ceria) in the platinum dispersion and the interaction between metal and support. Additionally, different Pt precursor are used to investigate if the properties mentioned above change by modifying the platinum precursor. To the best of our knowledge, specialized literature studying the effect of Pt precursor in catalysts is relatively scarce [40-43], especially those devoted to improving their performance in the DRM reaction. Thus, results are aimed to elucidate which of the characteristics (high metal dispersion or metal-support interaction) is relevant to the dry reforming of methane. The comparisons are performed using the same experimental conditions in both systems (synthesis and catalytic tests).
Experimental
Synthesis of materials
Commercial γ-Al2O3 from former Rhône Poulenc was used as alumina support. The sample was finely grounded and calcined at 800 °C for 4 h in a static air atmosphere; after calcination, the specific area was 172 m2 g-1, as shown in Table 1. On its side, CeO2 was prepared through a homogeneous precipitation method by urea hydrolysis, based on [44]. In a typical procedure, aqueous solutions of Ce(NO3)3·6H2O (Aldrich Cat. No. 238538) and (NH2)2CO (Sigma-Aldrich Cat. No.U5128) were mixed at room temperature. The initial pH value was in the range of 5-6 and it was controlled by adding small amounts of a HNO3 solution (EMD Cat. No. NX0412-2). Afterward, the solution was stirred for 12 h under reflux temperature (∼90 °C). The mixture was centrifuged, and the precipitate washed with deionized water and dried overnight. Finally, the solid was calcined at 800 °C for 4 h in a static air atmosphere.
Textural properties of as-prepared and spent catalysts.
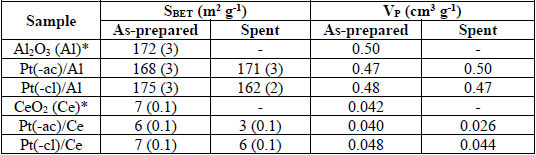
Pt catalysts (1.5 wt. %) were prepared via wet impregnation using two different platinum precursors, Platinum (II) acetylacetonate, Pt(acac)2 (Aldrich Cat. No. 282782) or Hexachloroplatinic acid, H2PtCl6·6H2O (Aldrich Cat. No. 206083), dissolved in acetone and deionized water, respectively. In both cases, appropriate amounts of the platinum solution were added dropwise to a stirring suspension containing the calcined support (Al2O3 or CeO2). The mixture was softly heated at 60 °C under agitation until complete evaporation of the solvent; afterward, solids were dried at 100 °C for 12 h. Fresh catalysts were first calcined in flowing air (30 mL min−1) at 500 °C for 2 h, and subsequently reduced in hydrogen flow of 10% H2/N2 (30 mL min−1) at 600 °C for 2 h; after this stage, materials are referred as as-prepared.
Catalysts are identified as Pt(-ac)/Ce and Pt(-ac)/Al for samples coming from the Pt(acac)2 precursor and as Pt(-cl)/Ce and Pt(-cl)/Al for those synthetized with the H2PtCl6·6H2O precursor; in both sets of catalysts, Ce and Al indicate the nature of the support, CeO2 and Al2O3, respectively.
Characterization of materials
The textural properties of as-prepared materials were characterized through N2 adsorption-desorption isotherms at -196 °C (N2-Ads) in a Quantachrome Autosorb MP-1 equipment. Before the experiments, samples (∼0.030 g) were degasified under vacuum at 200 °C. The specific area and pore volume were determined by the BET and BJH methods.
The crystalline structure of the phases present in the as-prepared materials was determined by powder X-ray diffraction (XRD). Experiments were performed at room temperature in a Bruker D8 diffractometer, using a CuKα radiation (λ = 0.154 nm), a filter of Ni-0.5%Cu-Kα in the secondary beam, and a 1- dimensional position sensitive silicon strip detector (Bruker, Linxeye). Measurements were carried out in the 20-120 ° range, with a 2θ step of 0.05 ° and 0.5 s per point. Determination of the average crystallite size and lattice parameter of materials was achieved by the Rietveld refinement of the XRD data, using the Fullprof code [45].
Transmission electron microscopy (TEM) was used to determine the size and structure of Pt species over as-prepared materials and the occurrence of carbon structures after the reaction. This twofold goal was achieved using a JEOL 2010 FEG microscope equipped with a Z-contrast annular (HAADF) detector and working at 200 kV. Materials were dry dispersed on commercial SPI lacey-carbon coated Cu grids for observations. The analysis of the images was performed using the Digital Micrograph 3.7.0 software.
The reduction properties of fresh and as-prepared materials were studied by temperature-programed reduction (H2-TPR), using a fixed-bed quartz reactor in a commercial reaction system coupled to a TCD detector (ISRI RIG-100). The reduction was performed under a flow of 5%H2/Ar (30 mL min-1) and a heating rate of 10 °C min-1 from room temperature up to 900 °C. The H2O produced by the reduction process was trapped before entered the TCD.
The dispersion of platinum in catalysts was determined from CO chemisorption experiments using the multitask ISRI RIG-100 reaction system. Before the characterization, as-prepared samples (0.100 g) were in-situ reduced at 400 °C for 1 h using a flow of 5%H2/He (30 mL min-1), then purged with helium (30 mL min-1) for 30 min and finally cooled down to room temperature; experiments were performed at this temperature. CO chemisorption took place when pulses of 0.5 mL (5%CO/He) were injected into a carrier stream of helium flowing at 30 mL min-1. The number of pulses must guarantee the formation of a CO monolayer chemisorbed over the Pt surface following a 1:1 stoichiometric ratio [46]. CO was monitored by a TCD detector coupled to the reaction system.
DRM reaction catalytic test
The DRM reaction was studied in a continuous flow fixed-bed quartz reactor under atmospheric pressure placed in a home-made dynamic reaction system coupled to a GOW-MAC HP 570 gas chromatograph equipped with a TCD detector and a Carboxen 1000 packed column to separate CO2, CH4, CO and H2 gas compounds. Before the tests, as-prepared samples (0.010 g) were in-situ reduced at 600 °C for 1 h using a 5%H2/He (30 mL min-1) flow. Afterward, samples were cooled down to 400 °C in helium for the activity tests, whereas for the stability experiments, the temperature was increased up to 800 °C in the helium atmosphere, after the reduction. In both approaches, the catalytic tests were performed using a reaction mixture with a volume composition of CO2:CH4:He = 1:1:8, and a total flow of 200 mL min-1 (WHSV = 85 h-1). Activity tests were carried out in the temperature range of 400-800 °C, with data collection each 50 °C and after the system reached steady state. On the other hand, stability tests were run at a fixed temperature (800 °C) for 24 h, collecting data each 0.2 h during this period.
Methane and carbon dioxide conversion (R(%)) were calculated as follows:
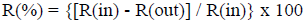
R stands for either the mole of methane or carbon dioxide admitted in the reactor (labeled as (in)) or measured in the effluent (labeled as (out)).
Additionally, the H2/CO molar ratio (SG) was obtained as follows:
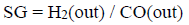
where, H2(out) and CO(out) are the mole of hydrogen and carbon monoxide produced in the reaction.
Characterization of spent materials
The characterization of materials after catalytic stability tests (800 °C / 24 h), labeled as spent, was performed via N2-Ads and TEM techniques, and using the same procedure as that described for the characterization of as-prepared catalysts.
Besides, the formation of carbonaceous species in the spent catalyst was determined using temperature-programed oxidation (O2-TPO). The study was performed in fixed-bed quartz reactor placed in a home-made dynamic reaction system coupled to a HIDEN HPR20 mass spectrometer. Prior to the experiments, samples (0.050 g) were treated at 200 °C in He flow for 1 h to remove residual adsorbed species. Oxidation occurs by the admission of a gas mixture flow of 20%O2/He (30 mL min-1) in the 200-800 °C interval and a heating rate of 10 °C min-1. The formation of CO2 was recorded following the m/e = 44 fragment.
Results and discussion
Physical characterization
N2 adsorption-desorption experiments characterized the textural properties of as-prepared materials. In all cases (supports and catalysts), the isotherms (not shown) are categorized as type IV, according to the IUPAC, which identifies mesoporous materials [47]. Depending on the nature of the support, isotherms present a different hysteresis loop: type H1 for alumina and type H3 for ceria.
The values of the specific area (SBET) and pore volume (VP) are displayed in Table 1. The calcined alumina exhibits a surface area around 170 m2 g-1 and after the impregnation of platinum, the textural properties of materials remain unaltered. It is worth noting that the SBET and VP values are significantly higher in Pt/Al2O3 samples.
The XRD patterns of as-prepared Pt/CeO2 catalysts are shown Fig. 1(a). All samples present the cubic fluorite structure of the CeO2 phase (JCPD PDF 43-1002). No crystalline platinum phases were identified, probably due to the low metal content (1.5 wt. %), below the technique detection limit. A good dispersion of the platinum species could also explain the absence of diffraction peaks. The structure parameters of ceria, calculated from the Rietveld refinement, are compiled in Table 2.
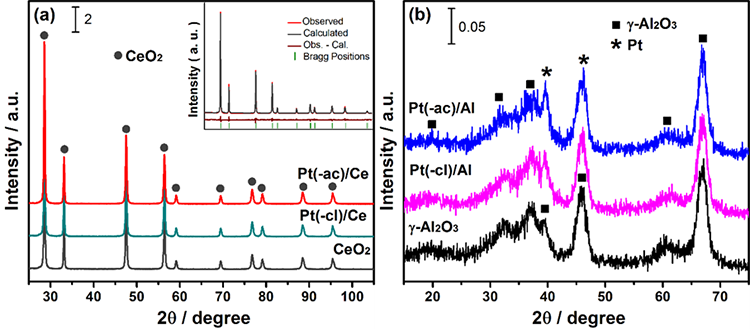
Fig. 1
XRD patterns of as-prepared catalysts: (a) Pt/CeO2 and (b) Pt/Al2O3. Inset in (a) displays an example of the Rietveld refinement.
CeO2 structure parameters of as-prepared Pt/CeO2 catalysts.
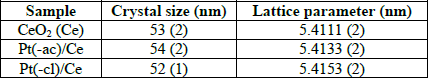
The mean crystallite size is around the same in all samples and remained unchanged after impregnation. The presence of these large ceria crystallites correlates well with the low specific area of the samples. In contrast, compared to the bare support, the lattice parameter of ceria increases after the impregnation of Pt. A lattice expansion takes place when a fraction of the ceria is reduced, as the ionic radii of Ce4+ (0.092 nm) and Ce3+ (0.103 nm) is different [48]. This process generates oxygen vacancies related to the oxygen storage capacity (OSC) of ceria. The enhanced oxygen storage capacity of ceria comes from the promoting effect of Pt, as reported elsewhere [49]. As the lattice expansion is more important in the Pt(-cl)/Ce sample, this is probably promoted by the presence of Cl species perturbing the ceria lattice [50,51].
On the other hand, the XRD patterns of as-prepared Pt/Al2O3 catalysts are displayed in Fig. 1(b). Diffractograms present broad peaks related to the poorly crystalline γ-Al2O3 phase (JCPD PDF 50-0741), making the refinement of the structure a complicated task.
After the addition of the platinum, no significant differences in the Al2O3 diffraction pattern are noticed. Nevertheless, some indication of signals related to the (111) and (200) planes of metallic Pt (JCPD PDF 04-0802) at around 39.7 and 46.2° overlapping with alumina peaks, are appreciated. Apparent observation of Pt phase only in the Pt/Al2O3 catalysts is probably related to differences in the crystallinity between the systems, impacting the scale of the diffraction patterns.
Characterization through STEM was carried out to determine the size and distribution of platinum particles over the as-prepared catalysts; representative images of ceria and alumina-supported samples are shown in Fig. 2. The bright spots in the images are related to Pt since in HAADF mode, the intensity is associated with the atomic number, higher in heavy atoms like platinum [52]. Table 3 shows the Pt average particle size in catalysts; dimensions were obtained through multiple micrographs and from at least 200 particles in each case. All catalysts present a reasonably small particle size, which indicates that the Pt species are well-dispersed, in agreement with the XRD characterization.
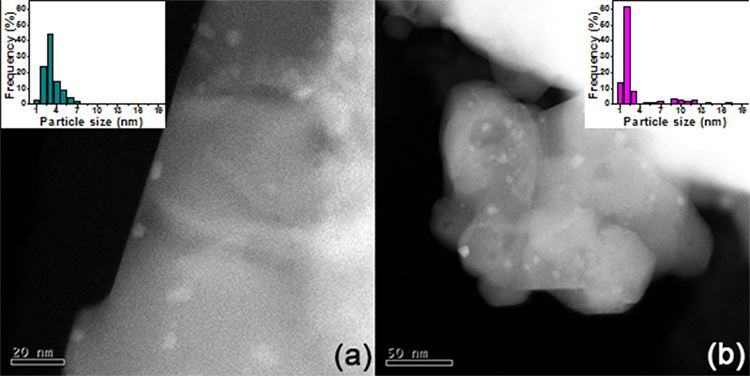
Fig. 2
Typical TEM-HAADF images and particle size histogram in the inset of as-prepared catalysts: (a) Pt(-cl)/Ce and (b) Pt(-cl)/Al.
Average particle size of Pt in as-prepared and spent catalysts (STEM); dispersion and particle size of reduced Pt catalysts (CO chemisorption).
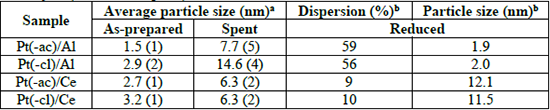
Despite the low specific area of Pt/CeO2 catalysts (Table 1), the average particle size of Pt is comparable to that of Pt/Al2O3 catalysts, showing that the metal dispersion is not only promoted by a high surface area, but also by a strong interaction with the support. Interestingly, regardless of the nature of the support, particle size is lower when Pt(acac)2 is used as the metal precursor.
Characterization of the structure of as-prepared catalysts was carried out through bright filed HRTEM. Representative images of ceria and alumina-supported catalysts are shown in Fig. 3. In both Pt(-ac)/Ce and Pt(-cl)/Ce catalysts, Figures 3(a)-(b), ceria and platinum species are discernible due to the differences in the size and contrast of the structures.
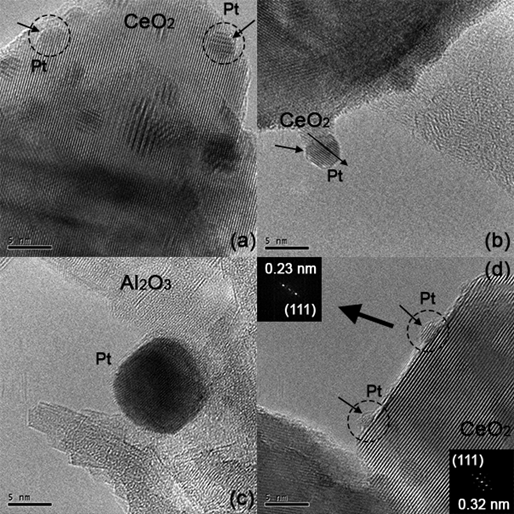
Fig. 3
Typical HRTEM images of as-prepared catalysts: (a) Pt(-ac)/Ce, (b) Pt(-cl)/Ce and (c) Pt(-ac)/Al. In (d) the FFT analysis of the structure of Pt and CeO2 in the Pt(-cl)/Ce sample.
Further analysis through fast Fourier transform (FFT) of the lattice fringes spacing in HRTEM images, Fig. 3(d), allows verifying the identity and exposed planes of CeO2 and Pt. Moreover, from examining the images, two features of Pt particles are recognized: i) a metal-support structural relationship (epitaxial growth), and ii) a covering of the Pt particles by an overlayer of a different phase.
A structural relationship between ceria and platinum particles is recognized by the parallel alignment of metal and support planes with the same Miller indexes [53]. The above is noticed in the micrographs (b) and (d) of Fig. 3 corresponding to Pt(-cl)/Ce catalyst; in the former an arrow passes through the in-plane alignment of CeO2 and Pt phases. Epitaxial relationships between Pt particles and ceria support are well documented [53-55]. For the second feature, i.e., Pt particles partially covered by a different phase, this is observed to some extent in the particles enclosed by circles, where differences, in contrast, are appreciated. This feature would be the manifestation of the classical SMSI effect observed in Metal/TiO2 catalysts [26], which in the case of CeO2 supported catalysts, the onset temperature is higher appearing above 500 °C [53]; as a recall, catalysts have been reduced at 600 °C.
On the other hand, a representative TEM image of alumina supported catalysts is displayed in Fig. 3(c), where the difference in the contrast allows the identification of the Pt particles.
Finally, the FFT-HRTEM analysis allowed discarding of other crystalline phases, like CeOCl (coming from the chloride precursor), or those formed with the support, as intermetallic Ce-Pt or platinum aluminate (PtAl2O4). The formation of the species mentioned above has been reported in other works with similar synthesis conditions [50,56]; thus, the confirmation of their absence limits materials catalytic activity to depend only on Pt and the supports.
Chemical characterization
The reduction properties of materials were studied through H2-TPR. Experiments performed over as-prepared catalysts (not shown) are characterized by an absence of H2 uptake in the 25-700 °C interval, and only those samples supported on ceria exhibit a peak above 700 °C, characteristic of the reduction of the CeO2 bulk [34]. The lack of reduction peaks shows the metallic state of platinum species in both systems, after the different thermal treatments (Section Synthesis of mateials) shown by XRD characterization, in Fig. 1(b) for Pt/Al2O3 samples. H2-TPR experiments over fresh samples were carried out to gain insight into the reducibility of the catalysts, and results are displayed in Fig. 4; for comparison purposes, the reduction profile of the corresponding calcined support is included. As a reminder, fresh samples are obtained after the drying step in the synthesis procedure and without further thermal treatments.
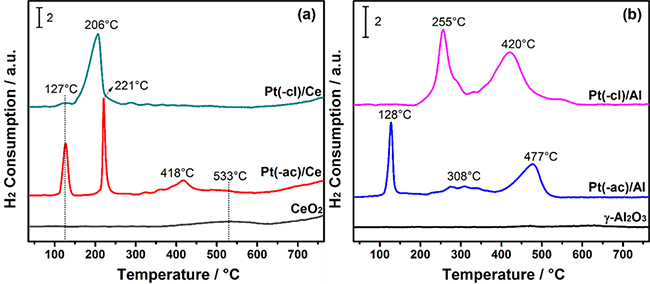
Fig. 4
H2-TPR profiles of fresh catalysts: (a) Pt/CeO2 and (b) Pt/Al2O3. In each system the reduction profile of the corresponding calcined support is included.
Differences in the TPR profiles are observed as a function of the Pt precursor and the support. For the fresh Pt/CeO2 catalysts, Fig. 4(a), the ceria support reduction features at high temperature (700 °C) seem unaltered, indicating that platinum interacts only with the CeO2 surface. The Pt(-cl)/Ce catalyst displays a tiny contribution around 125 °C and a broad H2 consumption centered at 206 °C, while in the Pt(-ac)/Ce sample, two intense sharp peaks are observed at 127 and 221 °C, along with a smaller peak at 418 °C. The Pt(-ac)/Ce catalyst also presents an ill-defined peak located around 530 °C, which matches the ceria surface reduction, identified in the support [34].
On their side, the H2-TPR profiles of fresh Pt/Al2O3 catalysts are shown in Fig. 4(b). The γ-Al2O3 support shows no hydrogen consumption, which agrees with the non-reducible nature of alumina [57]. Catalysts present different profiles depending on the platinum precursor. The Pt(-cl)/Al catalyst shows two broad hydrogen uptakes at 255 and 420 °C, while the Pt(-ac)/Al sample presents a sharp contribution at 128 °C, a flat peak around 308 °C and a broad H2 consumption at 477 °C.
The complexity of the profiles indicates that the reduction events arise from different origins. According to the literature, the thermal decomposition of H2PtCl6 and Pt(acac)2 in a hydrogen atmosphere occurs at moderate temperatures and produces mostly HCl and CH4, respectively [58]. Besides, the reduction of platinum (Pt4+ → Pt0) also consumes hydrogen in the process [59]. In this frame, an attempt to explain the differences in each sample will be made.
The single broad event in the Pt(-cl)/Ce sample (Fig. 4(a)), accounts for both the platinum reduction and the chloride precursor decomposition. The absence of the peak at ca. 530 °C (observed in the bare support) suggests that the reduction of the CeO2 surface is enclosed in the same event, and indicates an enhancement of the ceria reducibility. For the Pt(-ac)/Ce catalyst, the two sharp contributions are related to reducing platinum species interacting differently with ceria [60], while the peak around 418 °C characterizes the hydrocarbon species generated from the decomposition of acetylacetonate.
The peak around 255 °C in the Pt(-cl)/Al sample characterizes the reduction of platinum species while that at 420 °C is related to the decomposition of chloride species. In the Pt(-ac)/Al catalyst, the peak at 128 °C accounts for the platinum reduction while those around 308 and 477 °C identify the decomposition of the acetylacetonate precursor. Results indicate that the interaction of Pt with γ-Al2O3 is notably influenced by the precursor, modifying its reducibility.
Radivojević and co-workers [58] studied the thermal stability of various Pt precursors in hydrogen atmosphere and found that both H2PtCl6 and Pt(acac)2 precursors presented a similar behavior in terms of the temperature and path of decomposition. Thus, the different behavior of precursors decomposition (Fig. 4) is related to the interaction with the support.
For the samples prepared using Pt(acac)2, the decomposition of the precursor into hydrocarbon species in the Pt(-ac)/Ce sample is noticed by a single small peak at 418 °C, while for the Pt(-ac)/Al catalyst, this is noticed by two contributions, the one at 477 °C significantly more important. Similar observations in the H2-TPR profiles of Pt/Al2O3 catalysts obtained from acetylacetonate were found by Reyes and co-workers [43]. They report a complex profile featuring, among other things, important contributions located around 450 °C, ascribed to hydrogenation of carbonaceous fragments from the organometallic precursor.
On the other hand, in the Pt(-cl)/Ce sample, the close interaction between H2PtCl6 and ceria could lead to the simultaneous reduction of Pt and chlorinate species, while these two events appear separately in the Pt(-cl)/Al catalyst, as the interplay of the precursor with the γ-Al2O3 support is different.
The results of dispersion and particle size of Pt estimated by CO chemisorption are compiled in Table 3. Experiments were performed over as-prepared materials prior an in-situ thermal reactivation in hydrogen atmosphere. With this technique, platinum dispersion in Pt/Al2O3 catalysts (∼58 %) is higher than in the Pt/CeO2 catalysts (∼9 %). In both Pt(-ac)/Al and Pt(-cl)/Al samples, the particle size calculated from the dispersion values is about the same to that determined by STEM observations, which indicates that the in-situ reduction treatment of as-prepared samples does not promote particle sintering. Conversely, the particle size of the Pt(-ac)/Ce and Pt(-cl)/Ce catalysts is about six times higher compared to STEM one. For Pt/Al2O3 there is a good match between particle size values obtained from STEM and CO chemisorption techniques, which is not the case for Pt/CeO2.
The inconsistency between both values reveals that CO chemisorption is diminished in ceria supported catalyst after reduction at 600 °C. A lower chemisorbed CO amount traduces in low dispersion and larger particle size compared to STEM. As described in the introduction, a strong metal-support interaction (SMSI) like the one described by Tauster and co-workers [26] is likely to occur between Pt and CeO2 [46]. Partial blocking of the metal sites by reduced moieties of support would explain the lower amount of chemisorbed CO. As shown through the HRTEM characterization (Fig. 3), a covering of the Pt nanoparticles by other phase, presumable the reduced support, was evidenced. Both techniques, HRTEM and CO chemisorption sustain the hypothesis of a classical SMSI effect in the platinum-ceria system, and the differences in the particle sizes determined by STEM and chemisorption are ascribed to this interaction.
Catalytic properties in the DRM reaction
Before the full description of the catalytic properties, it is important to show the thermal treatments to which the fresh catalysts were submitted (Section Synthesis of materials) to choose the appropriate one. The selected procedure was based on the H2-TPR characterization of the fresh samples (Fig. 4), but additionally, by performing different activation processes and evaluating the catalytic performance in the DRM reaction. To illustrate the above, Table 4 shows the different temperatures in oxidizing and reducing environments used over the fresh Pt(-ac)/Ce sample and the corresponding methane conversion at 800 °C.
Thermal treatments tested in the Pt(-ac)/Ce catalyst and the corresponding activity in DRM reaction.

Results indicate that oxidation at 500 °C followed by a reduction at 600 °C leads to the higher activity (Table 4), probably by inducing a better interaction between the Pt and the support.
Activity test
The catalytic activity of in-situ reduced catalysts, in terms of the methane conversion in the range 400-800 °C is presented in Fig. 5(a). In the low-temperature regime (400-550 °C) no significant differences among the samples are observed while the methane conversion is almost negligible. At 600 °C, conversion reaches around 20 % in all samples, except the Pt(-ac)/Al catalyst. Finally, in the high-temperature regime (650-800 °C), the increment rate in the methane conversion follows the trend: Pt(-ac)/Ce > Pt(-cl)/Ce ≈ Pt(-cl)/Al > Pt(-ac)/Al.
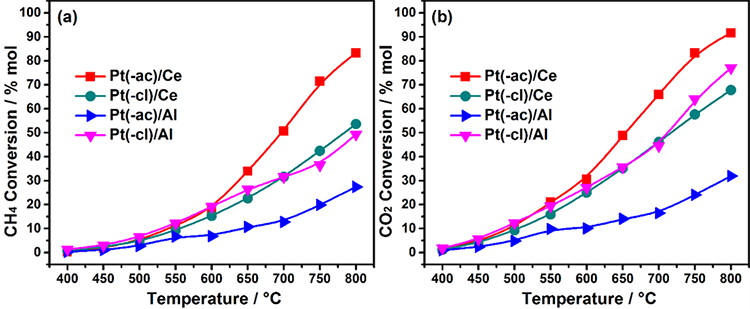
Fig. 5
Activity of in-situ reduced catalysts in the DRM reaction as a function of temperature in terms of: (a) Methane conversion and (b) Carbon dioxide conversion.
The Pt(-ac)/Ce catalyst performs better in the activity tests, indicating that CeO2 is a good option for platinum catalysts in the DRM reaction. Since the dispersion of Pt is about the same as in alumina supported catalysts (Table 3), the enhanced activity arises from the SMSI effect between platinum and ceria, as evidenced by the characterization (TEM, CO chemisorption). This strong interaction leads to more active sites, probably those at the interface issued from the Pt particle partial covering, where both adsorbed reactants activation occurs [61]. The lower activity in the Pt(-cl)/Ce sample could be related to residual chlorine species from the precursor that were not eliminated within the experimental conditions and were interfering with these rich interface sites. These entities are proposed to be different from the Cl species interacting closely with the ceria lattice and promoting the OSC (as evidenced by XRD).
According to the literature, the acidity of alumina (Brönsted sites) promotes the formation of carbonaceous species in the DRM reaction, which can be deposited over the Pt particle, the alumina support, or both [62-64], hence explaining the weak activity of these samples. However, the Pt(-cl)/Al catalyst activity is better, in contrast to its slightly larger Pt particle size (Table 3). This apparent discrepancy can be reconciled by considering that some residual chlorides from the precursor promote alumina acidity, which probably leads to the deposition of carbon on the support, rather than over the Pt particle. In that sense, the lower activity in Pt(-ac)/Al could also be related to higher deposition of carbonaceous deposits over the Pt sites. Overall, the effect of the platinum precursor takes relevance in the activity towards the DRM reaction.
CO2 conversion is higher in all samples, Fig. 5(b), which hints that other additional reactions are taking place. It has been reported that the DRM process involves the occurrence of side reactions like reverse water gas-shit (RWGS), Eq. (2), steam reforming of methane (SRM), Eq. (3), CH4 decomposition, Eq. (4), or CO disproportionation, Eq. (5) [3].
As it can be seen, Eq. (4) and (5) are a source of carbon, which deactivates the catalyst at different temperature regimes. Besides, in the presence of a reducible oxide support like CeO2, surface lattice oxygen could oxidize CO into CO2 [65].
 (2)
(2)
 (3)
(3)
 (4)
(4)
 (5)
(5)In line with the above, if only the DRM reaction occurs (Eq. (1)), the stoichiometric H2/CO molar ratio would be 1. This value is not attained by any catalyst (Fig. 6), proving that the reactions mentioned above (Eq. (2)-(5)) are also occurring, to some extent.
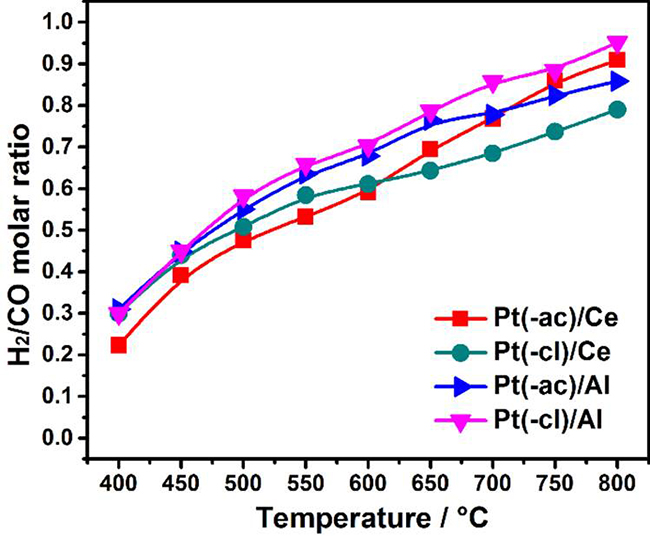
Fig. 6
H2/CO molar ratio displayed by in-situ reduced catalysts in the DRM reaction as a function of the reaction temperature.
The RWGS reaction occurrence explains both the increment in the CO2 conversion at high temperature and the lower H2/CO molar ratio. This ratio increases with the temperature, probably due to the SRM route (Eq. (3)), an endothermic reaction favored at high temperatures that consume the H2O produced by the RWGS reaction producing more H2.
Stability test
The catalytic stability in terms of the methane conversion for a 24 h on-stream run at 800 °C is displayed in Fig. 7(a) for in-situ reduced catalysts. Initially, the methane conversion in all samples is around 50-65 %; however, the stability is significantly different, depending on the support. This feature proves that not only the metal dispersion but also the support significantly impacts the stability of the Pt-based catalysts.
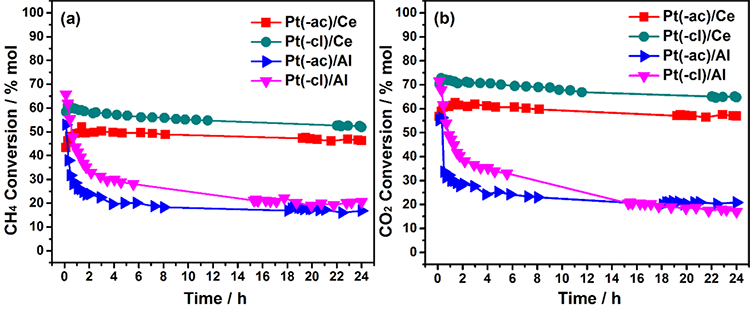
Fig. 7
Stability of in-situ reduced catalysts in the DRM reaction in terms of: (a) Methane conversion and (b) Carbon dioxide conversion.
The Pt/CeO2 catalysts present a slight diminution of the methane conversion along the run; in the end, the total drop is less than 5 %, indicating the excellent stability of these samples in the DRM reaction regardless the platinum precursor. This better performance, also observed in the activity tests (Fig. 5), is explained by the good metal dispersion and the presence of active interface sites resulting from the close interaction between Pt and ceria.
Although the stability is similar, the Pt(-cl)/Ce catalyst presents higher methane conversion along the run. This enhanced activity may be related to the metal-support structural relationship of platinum particles and the ceria support (Fig. 3(b)-(d)) along with the enhanced ceria reducibility (Fig. 4(a)), both factors probably leading to the improvement of the interface where the activation of reactants takes place [61]. Within the reaction conditions (800 °C), it seems that complete removal of residual chlorine species interfering with the interface sites is achieved, and thus their negative impact is avoided.
On the other hand, a quick deactivation within the first five hours on-stream (∼50 %) is noticed in both Pt/Al2O3 samples. Although the deactivation is slightly less pronounced in the Pt(-cl)/Al catalyst (following the activity tests), both exhibit a low methane conversion of about 20 % at the end of the run. This behavior shows that the stability of alumina-supported samples in the DRM reaction is poor, regardless of the platinum precursor.
As in the activity tests, both CH4 and CO2 conversion behave similarly (Fig. 7(b)); thus, the former discussion covers both. Nevertheless, a higher CO2 conversion is observed more importantly, in the Pt/CeO2 samples, which indicates that the parallel RWGS reaction takes place to a greater extent
Characterization of spent catalysts after stability tests
Different characterization techniques were applied to spent catalysts after the stability tests in the DRM reaction (800°C / 24 h) to correlate the structure features with the stability performance and elucidate the main deactivation route.
The determination of carbon deposits formed on the surface of spent catalysts was achieved through O2-TPO experiments, and results are displayed in Fig. 8. Profiles of Pt/Al2O3 samples show a broad CO2 production peak in the 300-600 °C interval, ascribed to gasification of different carbonaceous species, in the presence of oxygen (C + O2 → CO2) [18]. This peak is significant in the Pt(-cl)/Al sample, indicating that the amount of carbon deposits is higher in this catalyst. Results show that the formation of carbonaceous species in the DRM reaction is triggered by the γ-Al2O3 support and further promoted by the presence of residual chlorine.
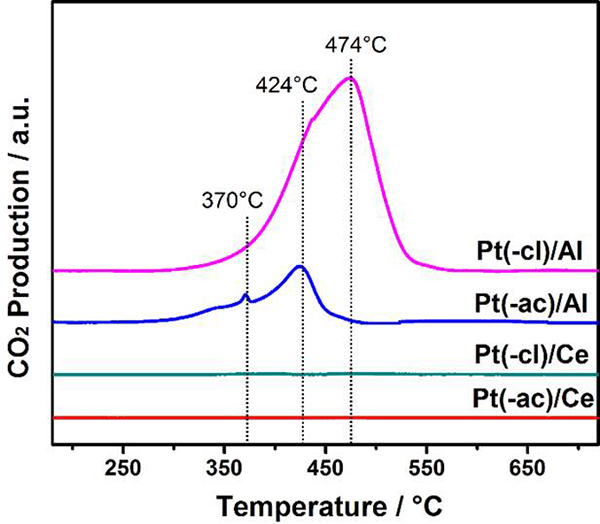
Fig. 8
O2-TPO profiles of spent catalysts after stability tests (800 °C / 24 h).
On the other hand, the CO2 signal in both Pt/CeO2 catalysts is negligible, which indicates that practically no carbon deposits blocking the active sites were generated during the DRM reaction, and correlates well with the excellent stability of these samples (Fig. 7). These anti-coking properties are related to the ability of CeO2 to switch from Ce4+ to Ce3+ while releasing oxygen, which either promotes in-situ carbon gasification or avoids its formation [35]. The promoting effect of Pt on this ceria property was evidenced by XRD characterization (Table 2, Section Physical characterization), more important in the Pt(-cl)/Ce sample, which agrees with its higher methane conversion.
The textural features of spent catalysts were studied via N2 physisorption. The isotherms of the spent Pt/Al2O3 samples (not shown) present about the same profile as that of as-prepared materials, while the SBET and VP values remain almost unaltered (Table 1), indicating the mesoporous structure of the γ-Al2O3 support is preserved and suggesting that deactivation issues in Pt/Al2O3 catalysts (Fig. 7) are not related to changes in the textural properties.
In contrast, the textural properties of the Pt/CeO2 system are more sensitive. The isotherms of spent catalysts (not shown) present changes in the hysteresis loop, while the pore size distribution determined by the BJH method indicates that the mesoporous structure collapses. The Pt(-ac)/Ce catalyst presents the most important diminution in both the SBET and VP values, Table 1. The lower methane conversion exhibited by this sample (Fig. 7) may be related to the diminution of the specific area in the hard-working conditions (DMR at 800 °C, 24 h). On the other hand, the partial covering of the Pt particle by the support is proposed to influence positively in the textural stability of the Pt(-cl)/Ce sample, thus improving the methane conversion.
Determination of the metal particle size in spent catalysts was achieved through HAADF-STEM analysis, and representative images are shown in Figures 9(a)-(b). The histograms (inset) reveal a broader particle size distribution compared to the as-prepared materials, Figures 2(a)-(b). An important increment in the particle size of Pt is observed in both Pt/Al2O3 catalysts, Table 3, however, the sintering is higher in the Pt(-cl)/Al sample, which suggests that the metal precursor impacts in the stability of the Pt species on alumina. The better stability of Pt particles average size in both Pt/CeO2 catalysts arise from the close interaction between the metal and the support and explains their better catalytic performance.
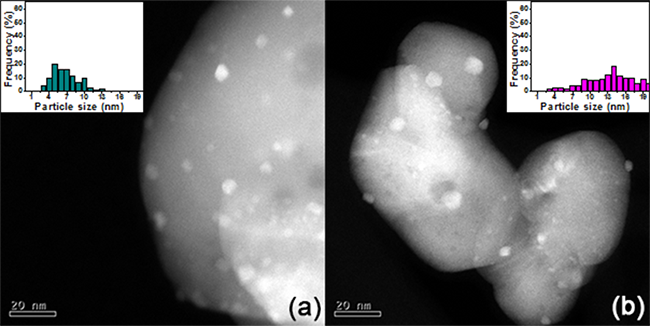
Fig. 9
Typical TEM-HAADF images of spent catalysts with particle size histogram in the inset: (a) Pt(-cl)/Ce and (b) Pt(-ac)/Al.
TEM was implemented for the identification of carbonaceous species in spent catalysts. Images of spent ceria-supported catalysts, Pt(-ac)/Ce and Pt(-cl)/Ce, are shown in Figures 10(a)-(b). No carbonaceous species are identified, while the Pt particle structure is preserved. Interestingly epitaxial growth of the Pt particle is still observed in the Pt(-cl)/Ce sample (Fig 10(b)). These results correlate well with the absence of CO2 production in the TPO experiments (Fig. 8) and evidence that carbon deposits are controlled in the Pt/CeO2 catalysts.
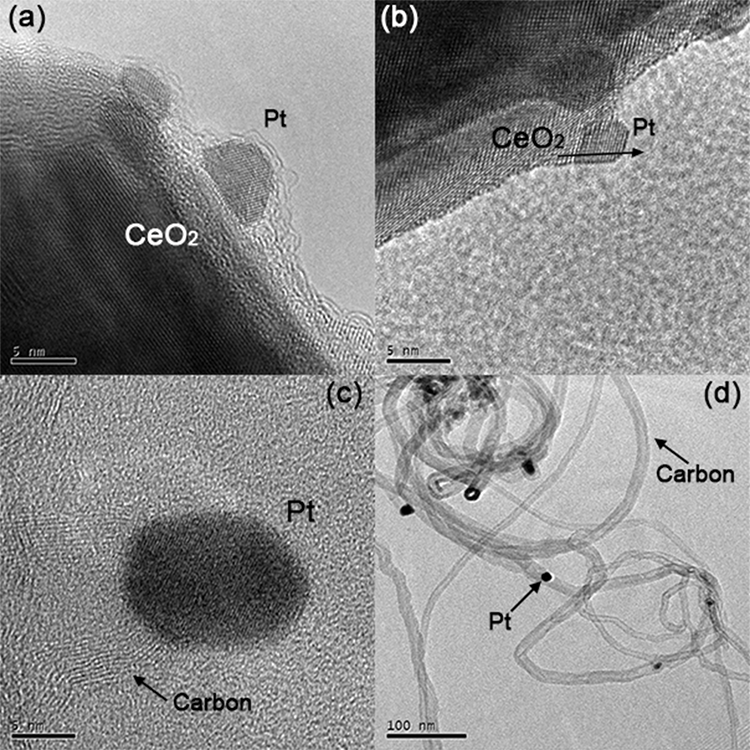
Fig. 10
Typical TEM images of spent catalysts: (a) Pt(-ac)/Ce, (b) Pt(-cl)/Ce, (c) Pt(-ac)/Al and (d) Pt(-cl)/Al.
Figures 10(c)-(d) display images of alumina-supported catalysts, Pt(-ac)/Al and Pt(-cl)/Al, where the formation of carbon filaments that occlude the Pt particles (darker spots) are observed. This encapsulation prevents the particle from contacting the reactants, and carbon structures production is proposed to be the main deactivation route for alumina-supported catalysts. According to the literature, these carbon filaments are generated by the methane decomposition (Eq. (4)), which is promoted at elevated temperatures; as the filament grows, it encapsulates the particle and detaches it from the support, deactivating the catalyst [66,67].
Conclusions
Platinum catalysts (1.5 wt. %) supported over CeO2 and γ-Al2O3 were successfully synthesized by wet-impregnation using two different precursors (Pt(acac)2 or H2PtCl6). Their catalytic properties were tested in the dry reforming of methane (DRM) reaction.
In general, characterization techniques revealed a comparable good platinum dispersion in both sets of supported ceria and alumina catalysts; however, the strong interaction of ceria with Pt led to a metal-support structural relationship and a partial covering of the Pt particles.
Both Pt/CeO2 catalysts performed better in the DRM reaction. The superiority of the ceria-supported catalysts was ascribed to the strong Pt-ceria interaction, leading to more active interface sites, lower Pt sintering, and carbon deposits suppression. The sample prepared with the Cl-containing platinum precursor presented increased ceria reducibility and oxygen mobility (OSC).
On their side, the effect of the precursor in the Pt/Al2O3 system was clearly noticed in the spent samples, as the Pt(-cl)/Al catalyst presented the higher amount of carbon deposits and the larger Pt particle sintering, both features responsible for the increased deactivation in the DRM reaction.
Overall, results contribute to the development of supported catalysts for DRM reaction as they evidenced that a strong interaction between the metal and the support is crucial in the better stability of Pt-based catalysts. This feature can be further tuned by selecting adequate support and metal precursor, and this approach could be applied in other metal-supported systems.
Acknowledgements
The authors thank M. Aguilar-Franco for the XRD characterization and L. Rendon for the TEM images. Daniel G. Araiza acknowledges CONACyT for the PhD scholarship No. 308329.
References
1. Cooper, S. A.; Raman, K. K.; Yin, J. J. Account. Public Policy 2018, 37, 226-240. https://doi.org/10.1016/j.jaccpubpol.2018.04.003
2. Song, C. Catal. Today 2006, 115, 2-32. https://doi.org/10.1016/j.cattod.2006.02.029
3. Abdulrasheed, A.; Jalil, A. A.; Gambo, Y.; Ibrahim, M.; Hambali, H. U.; Shahul Hamid, M. Y. Renew. Sustain. Energy Rev. 2019, 108, 175-193. https://doi.org/10.1016/j.rser.2019.03.054
4. Wender, I. Fuel Process. Technol. 1996, 48, 189-297.
5. Lunsford, J. H. Catal. Today 2000, 63, 165-174. https://doi.org/10.1016/S0920-5861(00)00456-9
6. Er-Rbib, H.; Bouallou, C.; Werkoff, F. Energy Procedia 2012, 29, 156-165. https://doi.org/10.1016/j.egypro.2012.09.020
7. Carapellucci, R.; Giordano, L. J. Power Sources 2020, 469, 228391. https://doi.org/10.1016/j.jpowsour.2020.228391
8. Arora, S.; Prasad, R. RSC Adv. 2016, 6, 108668-108688. https://doi.org/10.1039/c6ra20450c
9. Li, Z.; Wang, Z.; Kawi, S. ChemCatChem 2019, 11, 202-224. https://doi.org/10.1002/cctc.201801266
10. Singh, R.; Dhir, A.; Mohapatra, S. K.; Mahla, S. K. Biomass Convers. Biorefinery 2020, 10, 567-587. https://doi.org/10.1007/s13399-019-00417-1
11. Pakhare, D.; Spivey, J. Chem. Soc. Rev. 2014, 43, 7813-7837. https://doi.org/10.1039/c3cs60395d
12. Zhang, G.; Liu, J.; Xu, Y.; Sun, Y. Int. J. Hydrogen Energy 2018, 43, 15030-15054. https://doi.org/10.1016/j.ijhydene.2018.06.091
13. Kambolis, A.; Matralis, H.; Trovarelli, A.; Papadopoulou, C. Appl. Catal. A Gen. 2010, 377, 16-26. https://doi.org/10.1016/j.apcata.2010.01.013
14. Damyanova, S.; Pawelec, B.; Arishtirova, K.; Huerta, M. V. M.; Fierro, J. L. G. Appl. Catal. B Environ. 2009, 89, 149-159. https://doi.org/10.1016/j.apcatb.2008.11.035
15. García-Diéguez, M.; Finocchio, E.; Larrubia, M. Á.; Alemany, L. J.; Busca, G. J. Catal. 2010, 274, 11-20. https://doi.org/10.1016/j.jcat.2010.05.020
16. Seo, H. G.; Ji, S.; Seo, J.; Kim, S.; Koo, B.; Choi, Y.; Kim, H.; Kim, J. H.; Kim, T. S.; Jung, W. C. J. Alloys Compd. 2020, 835, 155347. https://doi.org/10.1016/j.jallcom.2020.155347
17. Nagai, Y.; Dohmae, K.; Ikeda, Y.; Takagi, N.; Hara, N.; Tanabe, T.; Guilera, G.; Pascarelli, S.; Newton, M. A.; Takahashi, N.; Shinjoh, H.; Matsumoto, S. Catal. Today 2011, 175, 133-140. https://doi.org/10.1016/j.cattod.2011.02.046
18. Araiza, D. G.; Arcos, D. G.; Gómez-Cortés, A.; Díaz, G. Catal. Today 2019, No. In Press. https://doi.org/10.1016/j.cattod.2019.06.018
19. Zhang, Q.; Long, K.; Wang, J.; Zhang, T.; Song, Z.; Lin, Q. Int. J. Hydrogen Energy 2017, 42, 14103-14114. https://doi.org/10.1016/j.ijhydene.2017.04.090
20. Schubert, M. M.; Hackenberg, S.; Van Veen, A. C.; Muhler, M.; Plzak, V.; Behm, J. J. J. Catal. 2001, 197, 113-122. https://doi.org/10.1006/jcat.2000.3069
21. Das, S.; Sengupta, M.; Patel, J.; Bordoloi, A. Appl. Catal. A Gen. 2017, 545, 113-126. https://doi.org/10.1016/j.apcata.2017.07.044
22. Jayabal, S.; Saranya, G.; Geng, D.; Lin, L. Y.; Meng, X. J. Mater. Chem. A 2020, 8, 9420-9446. https://doi.org/10.1039/d0ta01530j
23. Adamiec, J.; Fiedorow, R. M. J.; Wanke, S. E. J. Catal. 1985, 95, 492-500. https://doi.org/10.1016/0021-9517(85)90127-7
24. van Deelen, T. W.; Hernández Mejía, C.; de Jong, K. P. Nat. Catal. 2019, 2, 955-970. https://doi.org/10.1038/s41929-019-0364-x
25. Wu, P.; Tan, S.; Moon, J.; Yan, Z.; Fung, V.; Li, N.; Yang, S. Z.; Cheng, Y.; Abney, C. W.; Wu, Z.; Savara, A.; Momen, A. M.; Jiang, D. en; Su, D.; Li, H.; Zhu, W.; Dai, S.; Zhu, H. Nat. Commun. 2020, 11, 1-10. https://doi.org/10.1038/s41467-020-16674-y
26. Tauster, S. J.; Fung, S. C.; Baker, R. T. K.; Horsley, J. A. Science (80-. ). 1981, 211, 1121-1125. https://doi.org/10.1126/science.211.4487.1121
27. Li, S.; Xu, Y.; Chen, Y.; Li, W.; Lin, L.; Li, M.; Deng, Y.; Wang, X.; Ge, B.; Yang, C.; Yao, S.; Xie, J.; Li, Y.; Liu, X.; Ma, D. Angew. Chemie - Int. Ed. 2017, 56, 10761-10765. https://doi.org/10.1002/anie.201705002
28. Zhao, E. W.; Zheng, H.; Ludden, K.; Xin, Y.; Hagelin-Weaver, H. E.; Bowers, C. R. ACS Catal. 2016, 6, 974-978. https://doi.org/10.1021/acscatal.5b02632
29. Chen, B.; Zhao, Q.; Yu, L.; Chen, L.; Crocker, M.; Shi, C. Catal. Sci. Technol. 2020, 10, 4571-4579. https://doi.org/10.1039/d0cy00857e
30. Bonneviot, L.; Haller, G. L. J. Catal. 1991, 130, 359-373. https://doi.org/10.1016/0021-9517(91)90120-S
31. Trueba, M.; Trasatti, S. P. Eur. J. Inorg. Chem. 2005, No. 17, 3393-3403. https://doi.org/10.1002/ejic.200500348
32. Borgna, A.; Garetto, T. F.; Apesteguía, C. R.; Le Normand, F.; Moraweck, B. J. Catal. 1999, 186, 433-441. https://doi.org/10.1006/jcat.1999.2557
33. García-Diéguez, M.; Pieta, I. S.; Herrera, M. C.; Larrubia, M. A.; Malpartida, I.; Alemany, L. J. Catal. Today 2010, 149, 380-387. https://doi.org/10.1016/j.cattod.2009.07.099
34. Trovarelli, A. Catal. Rev. 1996, 38, 439-520. https://doi.org/10.1080/01614949608006464
35. Araiza, D. G.; Gómez-Cortés, A.; Díaz, G. Catal. Today 2020, 349, 235-243. https://doi.org/10.1016/j.cattod.2018.03.016
36. Rodas-Grapaín, A.; Arenas-Alatorre, J.; Gómez-Cortés, A.; Díaz, G. Catal. Today 2005, 107-108, 168-174. https://doi.org/10.1016/j.cattod.2005.07.167
37. Yang, M.; Guo, H.; Li, Y.; Dang, Q. J. Nat. Gas Chem. 2012, 21, 76-82. https://doi.org/10.1016/S1003-9953(11)60336-8
38. Nagai, Y.; Hirabayashi, T.; Dohmae, K.; Takagi, N.; Minami, T.; Shinjoh, H.; Matsumoto, S. J. Catal. 2006, 242, 103-109. https://doi.org/10.1016/j.jcat.2006.06.002
39. Jiang, S. P. Mater. Sci. Eng. A 2006, 418, 199-210. https://doi.org/10.1016/j.msea.2005.11.052
40. Claudio-Piedras, A.; Ramírez-Zamora, R. M.; Alcántar-Vázquez, B. C.; Gutiérrez-Martínez, A.; Modragón-Galicia, G.; Morales-Anzures, F.; Pérez-Hernández, R. Catal. Today 2019, No. July. https://doi.org/10.1016/j.cattod.2019.08.013
41. Hu, L.; Boateng, K. A.; Hill, J. M. J. Mol. Catal. A Chem. 2006, 259, 51-60. https://doi.org/10.1016/j.molcata.2006.06.018
42. Marceau, E.; Lauron-Pernot, H.; Che, M. J. Catal. 2001, 197, 394-405. https://doi.org/10.1006/jcat.2000.3078
43. Reyes, P.; Oportus, M.; Pecchi, G.; Fréty, R.; Moraweck, B. Catal. Letters 1996, 37, 193-197. https://doi.org/10.1007/BF00807753
44. Matijević, E.; Hsu, W. P. J. Colloid Interface Sci. 1987, 118, 506-523. https://doi.org/10.1016/0021-9797(87)90486-3
45. Rodríguez-Carvajal, J. Phys. B Phys. Condens. Matter 1993, 192, 55-69. https://doi.org/10.1016/0921-4526(93)90108-I
46. Holmgren, A.; Andersson, B.; Duprez, D. Appl. Catal. B Environ. 1999, 22, 215-230. https://doi.org/10.1016/S0926-3373(99)00047-8
47. Thommes, M.; Kaneko, K.; Neimark, A. V.; Olivier, J. P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K. S. W. Pure Appl. Chem. 2015, 87, 1051-1069. https://doi.org/10.1515/pac-2014-1117
48. Zhang, F.; Chan, S. W.; Spanier, J. E.; Apak, E.; Jin, Q.; Robinson, R. D.; Herman, I. P. Appl. Phys. Lett. 2002, 80, 127-129. https://doi.org/10.1063/1.1430502
49. Bera, P.; Priolkar, K. R.; Gayen, A.; Sarode, P. R.; Hegde, M. S.; Emura, S.; Kumashiro, R.; Jayaram, V.; Subbanna, G. N. Chem. Mater. 2003, 15, 2049-2060. https://doi.org/10.1021/cm0204775
50. Fajardie, F.; Tempere, J. F.; Manoli, J. M.; Djega-Mariadassou, G.; Blanchard, G. J. Chem. Soc. - Faraday Trans. 1998, 94, 3727-3735. https://doi.org/10.1039/a805625k
51. Liu, J.; Hao, M.; Chen, C.; Du, K.; Zhou, Q.; Zou, S.; Xiao, L.; Fan, J. Appl. Surf. Sci. 2020, 528, 147025. https://doi.org/10.1016/j.apsusc.2020.147025
52. Pennycook, S. J. Ultramicroscopy 1989, 30, 58-69. https://doi.org/10.1016/0304-3991(89)90173-3
53. Bernal, S.; Calvino, J. J.; Cauqui, M. A.; Gatica, J. M.; Larese, C.; Pérez Omil, J. A.; Pintado, J. M. Catal. Today 1999, 50, 175-206. https://doi.org/10.1016/S0920-5861(98)00503-3
54. Penner, S.; Wang, D.; Podloucky, R.; Schlögl, R.; Hayek, K. Phys. Chem. Chem. Phys. 2004, 6, 5244-5249. https://doi.org/10.1039/b410124c
55. Datye, A. K.; Kalakkad, D. S.; Yao, M. H.; Smith, D. J. Journal of Catalysis. 1995, pp 148-153. https://doi.org/10.1006/jcat.1995.1196
56. Hwang, C. P.; Yeh, C. T. J. Mol. Catal. A Chem. 1996, 112, 295-302. https://doi.org/10.1016/1381-1169(96)00127-6
57. Melchor-Hernández, C.; Gómez-Cortés, A.; Díaz, G. Fuel 2013, 107, 828-835. https://doi.org/10.1016/j.fuel.2013.01.047
58. Radivojević, D.; Seshan, K.; Lefferts, L. Appl. Catal. A Gen. 2006, 301, 51-58. https://doi.org/10.1016/j.apcata.2005.11.016
59. Lin, W.; Herzing, A. A.; Kiely, C. J.; Wachs, I. E. J. Phys. Chem. C 2008, 112, 5942-5951. https://doi.org/10.1021/jp710591m
60. Zhou, A.; Wang, J.; Wang, H.; Li, H.; Wang, J.; Shen, M. J. Rare Earths 2018, 36, 257-264. https://doi.org/10.1016/j.jre.2017.07.008
61. Yeung, C. M. Y.; Yu, K. M. K.; Fu, Q. J.; Thompsett, D.; Petch, M. I.; Tsang, S. C. J. Am. Chem. Soc. 2005, 127, 18010-18011. https://doi.org/10.1021/ja056102c
62. Querini, C. A.; Fung, S. C. Catal. Today 1997, 37, 277-283. https://doi.org/10.1016/S0920-5861(97)00020-5
63. Shamsi, A.; Baltrus, J. P.; Spivey, J. J. Appl. Catal. A Gen. 2005, 293, 145-152. https://doi.org/10.1016/j.apcata.2005.07.002
64. Lercher, J. A.; Bitter, J. H.; Hally, W.; Niessen, W.; Seshan, K. Stud. Surf. Sci. Catal. 1996, 101 A, 463-472. https://doi.org/10.1016/s0167-2991(98)80284-x
65. Wu, Z.; Li, M.; Overbury, S. H. J. Catal. 2012, 285, 61-73. https://doi.org/10.1016/j.jcat.2011.09.011.
66. Snoeck, J. W.; Froment, G. F.; Fowles, M. J. Catal. 1997, 169, 240-249. https://doi.org/10.1006/jcat.1997.1634.
67. Li, Y.; Li, D.; Wang, G. Catal. Today 2011, 162, 1-48. https://doi.org/10.1016/j.cattod.2010.12.042
Author notes
*Corresponding author: Daniel G. Araiza, email [email protected]; Gabriela Díaz, email [email protected]
