Journal of the Mexican Chemical Society, vol. 63, no. 4, 2019
Sociedad Química de México A.C.
Rahma Mahrous
Alexandria University, Egypt
Hoda M. Fathy *
Alexandria University, Egypt
Rasha M. Abu El-Khair
Alexandria University, Egypt
Abdallah A. Omar
Alexandria University, Egypt
Received: 13 June 2019
Accepted: 04 September 2019
Abstract.: Seven compounds were isolated from the leaves and fruits of Egyptian Withania somnifera dunal, (Family: Solanaceae). The identity of the compounds based on their spectroscopic data were as follows: two withanolides; withaperuvin C (1), phyperunolideF (2) and four lipids;1,2-di-O-palmitoyl-3-O-(6´´´-sulfo-α-D-quinovopyranosyl)-glycerol (3), vaccenic acid (5), 1,3 dicaproyl,2-vaccenoyl-glycerol (6), vaccenolymonoglyceride (7) and β-sitosterol glucoside (4). All the isolated metabolites except (4) were reported for the first time from this plant. Besides, this is the first report for isolation of compounds (6, 7) in a pure form from a natural source. Different fractions of the fruits of the Egyptian plant were investigated for their anticholinesterase activities where the most potent ones found to be the aqueous, the light petroleum fraction in addition to a mixture of lipids.
Keywords: C, phyperunolide, vaccenoylmonoglyceride, anticholinesterase, withania.
Resumen.: Se aislaron siete compuestos de las hojas y frutos del dunal egipcio Withania somnifera, (Familia: Solanaceae). La identidad de los compuestos, basada en sus datos espectroscópicos, fue la siguiente: dos witanólidos; witaperuvina C (1), fiperunolida F (2) y cuatro lípidos;1,2-di-O-palmitoil-3-O-(6´´´-sulfo-α-D-quinovopiranosil)-glicerol (3), ácido vacénico (5), 1,3 dicaproil, 2-vacenoil-glicerol (6), vacenolimonoglicérido (7) and β-sitosterol glucósido (4). Todos los metabolitos aislados de esta planta, excepto (4), son reportados por primera vez. Además, este es el primer informe para el aislamiento de compuestos (6, 7) en forma pura a partir de una fuente natural. Se investigaron diferentes fracciones de los frutos de la planta por su actividad anticolinesterasa, siendo las más potentes la fracción acuosa, la fracción ligera de petróleo, además de una mezcla de lípidos.
Palabras clave: Witaperuvina C, fiperunólida, vacenoilmonoglicérido, anticolinesterase, witania.
Introduction
Withania somnifera (L)Dunal, commonly known as Ashwagandha, is one of the most valued medicinal plants in Ayurveda that is used as a general tonic. The ethnopharmacological properties of the plant include adaptogenic, anti-sedative and anti-convulsion activities. The plant is also used to treat various neurological disorders, arthritis, stress and behavior-related problems[1-4]. Although the plant is widely distributed in Egyptian flora, few studies have been carried out to explore its constituents and biological activities, compared to the Indian one. Our previous investigation showed that ethanolic extracts of all the plant parts had significant cytotoxic activity against liver cancer cells with the highest activity manifested by the leaves extract[5].Our previous study of the leaves led to the isolation of several withanolides mainly withanolide E, and withanolide S. which is significantly different from the Indian plant in the chemistry of the isolated withanolides[5]. The Indian plant is known for the presence of withaferin A and withanolideA representing its main components [6,7]. These withanolides were not detected in the Egyptian plants while 20 hydroxywithanolides with α -oriented side chain were present as reported in our earlier study [8]. The occurrence of these particular withanolides in their respective ratios in the leaf extract showed that the Egyptian plant belongs to chemotype III [5].
In continuation of our interest in the Egyptian plant, we demonstrated the anticholinesterase activity manifested by all the plant parts with the highest one produced by the ripe fruit extracts [8]. The extracts showed also significant antioxidant, nitric oxide scavenging and anti-inflammatory activities. Withanolide S, isolated from the leaf extract, exhibited strong in vitroacetylcholinesterase (AChE) inhibitory activity, with an IC50 value of 0.00035 μM [8]. Our earlier work concluded that, unlike the Indian plant that is reputed for the medicinal use of its roots, all the plant parts extracts of Egyptian W. somnifera showed significant anticholinesterase activity.
In this study, we considered the anticholinesterase activity of different fractions of the ripe fruits of the Egyptian plant aiming to explore its chemical constituents and isolate some metabolites that may further contribute to the already known biological activities regarding Alzheimer's disease. In addition to further phytochemical investigation of the leaves extract.
Experimental
General experimental procedure
The 1D and 2D NMR analyses were obtained using a Brukeravance III (Switzerland) 400 MHz. Residual peaks of the deuterated solvents were used to reference the spectrum. ESIMS spectra were measured on Waters Xevo G2-S Q-TOF mass spectrometer (Milford, MA, USA).
Acetylthiocholine iodide (ACTI), 5,5′-dithiobis[2-nitrobenzoic acid] (DTNB),diphenyl-1-picrylhydrazyl (DPPH) and sulphanilamide were purchased from Sigma-Aldrich ,USA. N Silica gel (230-400 mesh), precoated TLC plates silica gel 60 (GF-254) were purchased from Merck, Germany. All the solvents used were of analytical grade.
Instrumentation and software
Plant material
Withania somnifera (L) Dunal was collected from Alexandria, Egypt and described in detail in our previous publications [8]
Extraction and isolation of compounds (1-4) from leaves extract
The dried powdered leaves (600 g) were defatted using light petroleum, the defatted powder was air dried then extracted with 75% ethanol. The dried ethanolic extract (40 g) was re-dissolved in 70% ethanolthen extracted successively with light petroleum. The lower layer was concentrated and then partitioned between 60% ethanol and methylene chloride. The later (11.2 g) was subjected to a silica gel column chromatography packed in methylene chloride. Elution was carried with increasing concentrations of ethyl acetate in methylene chloride, followed by methanol. Theeluted fractionwith 60% ethyl acetate in methylene chloride (5 g) was further separated by silica gel column using ethyl acetate - methylene chloride (1:1) to afford compounds 1 and 2(10 mg each). Fraction eluted with 5% methanol in ethyl acetate yielded compound 3 (60 mg) as needle shaped crystals. Fraction eluted with 60% methanol in ethyl acetate yielded compound 4 (30 mg) as an amorphous powder.
Isolation of compounds (5-7) from ripe fruits extract
The powdered air-dried ripe fruits (500g) were macerated with light petroleum (60-80), filtered ,air dried, then extracted with 70% ethanol .The light petroleum extract of ripe fruits was evaporated to dryness to give an orange semisolid residue (34g) then it was re-dissolved in 500ml light petroleum and partitioned with 90% ethanol. The hydroalcoholic layer was separated and while concentrating a mixture of fatty nature that afforded three major spots “WR” was precipitated and purified using column chromatography and elution was carried with hexane with increasing concentration of methylene chloride to give compound 5 (80 mg), compound 6 (20 mg), compound 7 (5 mg).
The dried alcoholic extract of the ripe fruits (30g) was re-dissolved in (500ml 70% ethanol) and successively extracted with methylene chloride, ethyl acetate and n- butanol.
Hydrolysis of glycerides (compound 6 and 7)
Ten mg of the triglyceride were refluxed for one hour with 1M potassium hydroxide in 95% ethanol and the free fatty acids were recovered after acidification.
Acetylcholinesterase activity
Six Balb/c mice were obtained from the animal house of the Medical Research Institute, Alexandria University. After anaesthesia, six brains were isolated and washed in cold saline. One gram of the brains was homogenized in 9 ml phosphate buffer saline. The homogenate was centrifuged at 3000 rpm and the supernatant was used for further biochemical estimations. The experimental protocol was approved by the Animal Care & Use Committee (ACUC), Faculty of Pharmacy, Alexandria University, Egypt and was given number (AUC17/12).
Acetylcholinesterase (AChE) activity was assessed according to the method of Ellmanet al[10] in which 130 μl phosphate buffer (0.1 M pH 7.4) were added to a mixture of 20 μl brain homogenate and 20 μl of different concentration of each extract (test) or organic solvent (control), then incubated for 45 min at 37°C. Five μl of substrate Acetylthiocholine iodide ACTI (75 mM) were added, mixed well and incubated for 15 min at 37°C. Finally, 60 μl 5,5-dithiobis[2-nitrobenzoic acid] DTNB (0.32 mM) were added and left for 5 min. The absorbance was measured at 405 nm and the specific activity was calculated using the following equations.
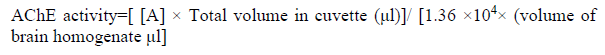 (1)
(1)
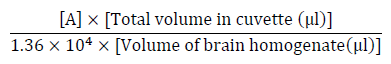 (2)
(2)
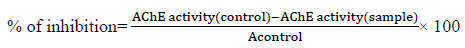 (3)
(3)where, 1.36 X 104 is the molar extinction coefficient of DTNB.
All the dried extracts and fractions were dissolved in dimethylsulfoxide (DMSO) and stock solutions of the extracts were appropriately diluted to get different concentrations (0.2-1 mg/ml) for each extract/ fraction and were used for biological analysis.
Statistical analysis
All experiments were conducted in triplicate and values expressed as mean ± standard deviation, correlations were carried out using the correlation and regression in the EXCEL program.
Spectral data
Withaperuvin C(1)
An off-white amorphous powder freely soluble in methanolwith a melting point of 190 ° C. UV λ max (MeOH) 312 nm.C28H38O7, 1H NMR (CDCl3, 500 MHz): δH (ppm)6.99 (1H, br s, H-3), 5.97 (1H, J= 12 Hz, H-2 )5.79 (1H, s, H-4 ),4.71 (1H, H-22) , 4.64 (1H, d, J=8Hz, H-6̀) , 2.39 (2H,m, H-12b, H-7b) ,: 2.35 (2H , H-12a, H-8) , 2.08 (1H,m, H-15b) , 1.90 (3H,s, H-28), 1.76 (4H, s, H-27, H-16)1.66 (2H, H-23b , H-9) ,H-23a), 1.40 (1H) , 1.52 (2H, m, H-16a, H-11, H-7a), 1.24 (3H, s, H-21), 1.12 (3H, s, H-19), 0.98 (3H, s, H-18), and 13CNMR (CDCl3, 125 MHz , assignment by APT and HMQC):δC (ppm):205.1 (C, C-1), 166.4 (C,C-26), 160.2 (C, C-5), 151.4 (C, C-24), 141.5 (CH, C-3), 125.6 (CH, C-4), 120.5 (C, C-24), 116.6 (CH,C-2),87.5 (C, C-17), 82.7 (C, C-14), 81.4 (CH,C-22) , 78.6 (C,C-20), 73.0 (CH,C-6), 54.2 (C, C13, C-10), , 42.93 (CH, C-9), 37.5 (CH2, C-16), 36.3 (CH2, C-7), 34.7 (CH2, C-12), 34.59 (CH, C8), 30.4 (CH2, C-15), 22.0 (CH2, C-11), 32.7 (CH2, C-23), 20.8 (CH3, C-18), 20.9 (CH3, C-19) , 20.8 (CH3, C-28), 19.6 (CH3, C-21), 12.6 (CH3, C-27).
Phyperunolide F (2)
Off-white amorphous powder freely soluble in methanol. 1H NMR (CDCl3, 500 MHz):δH (ppm)3.83 (1H, s, H-4), 3.45 (1H, br s, H-3), 3.16 (1H, s, H-6), 2.75 (1H, dd, J =8Hz, 4Hz H-2b), 2.56 (1H, d, J=8Hz, H-2a), 2.28 (1H, s,H-8),1.85 (2H, d, J=4Hz, H-7), 1.81 (1H, d, J=4Hz, H-9), 1.25* (1H, H-11a), 1.37 (1H, m, H-11b) , 1.25* (1H, H-12a), 2.13(1H, m, H-12b), 1.40 (1H, s, H-15a), 1.54# (1H, d, J=8Hz , H-15b), 1.43 (1H, s, H-16a), 1.54# (1H, d, J=8Hz, H-16b), 1.25* (3H, H-18), 1.07$ (3H, t, J=16, 8, 4Hz, H-19), 0.93 (3H, s, H-21), 4.63 (1H, dd, J=12Hz, 4Hz, H-22), 2.41 (1H, br s, H-23a), 2.44 (1H, br s, H-23b), 1.75 (3H,s, H-27), 1.88 (3H,s, H-28), 3.39 (2H, br s, H-1´), 1.07$ (3H, t, J=16, 8, 4Hz, H-2´ ) and 13CNMR (CDCl3, 125 MHz, assignment by APT and HMQC):δC (ppm):211 (C, C-1), 166.4 (C,C-26), 151.4 (C,C-24), 120.4 (C,C-25), 87.6 (C,C-17), 72.9 (CH, C-3), 81.6 (C,C-14), 70.8 (CH, C-4), 63.0 (CH2, C-1̀), 81.3 (CH, C-22), 78.7 (C,C-20), 62.3 (C,C-5), 61.9 (CH, C-6), 43.1 (CH2,C2), 51.69 (C,C-10), 54.2 (C,C-13), 34.7, 35.4 (CH, C-9), 33.4 (CH, C-8), 36.3 ( CH2, C-16), 26.4 ( CH2, C-7), 29.7 ( CH2, C-12), 32.4 ( CH2, C-15), 15.8 (CH3, C-2̀), 22.29 ( CH2, C-11), 19.5( CH3, C-18), 20.6 ( CH3, C-21), 15.8 (C,C-19), 20.7 (CH3, C-28), 12.6 (CH3, C-27).
1,2-di-O-palmitoyl-3-O-(6´´´-sulfo-α-D-quinovopyranosyl)-glycerol (3)
The IR spectra 3459 2919, 2851 1736 & 1064, 1215 & 1064 ,794 and 720. ESIMS m/z (% int.): 793.8 (100%) [M -H]- calculated for C41H77O12SH. 1H-NMR (DMSO-d6, 500 MHZ):δH (ppm)5.14 (1H, d, J=5.3Hz, H-2), 4.58 (1H, s, H-1´´´)4.36 (1H, d, J=12Hz H-1a), 4.15 (1H, q, J=12, 5Hz H-1b) , , 3.79(1H, brs, H-5´´´) 3.34 (H-3b partially masked by the water peak) ,3.33 (H-3´´´partially masked by the water peak), 2.93 (1H, br s, H-4´´´) , 2.86 (1H, d, H:-6´´´ a), 2.58(1H, d H-6´´´ b), 2.27 (4H, m, H-2´, 2´´), 1.5 (4H, s, H-3´, 3´´), 1.24 (48H, br s, -CH2 of fatty acid), 0.86 (6H, s, 16´,16´´, CH3)and 13CNMR (DMSO-d6, 125 MHz, , assignment by APT and HMQC):δC (ppm):172.82, 172.98 (C, C-1´,1´´), 98.8 (CH, C-1´´´),74.7 (CH, C-4´´´), 73.37 (CH, C-3´´´), 72.09 (CH, C-2´´´) , 70.2 (CH, C-2), 69.02 (CH, C-5´´´), 65.12 (CH2, C-3), 63.1 (CH2, C-1), 55.066 (CH2, C6´´´), 33.9, 34.04 (CH2, C2´,2´´), 24.93 (CH2, C-3´,3´´), 31.78- 22.57 (-CH2 of fatty acid) , 14.39 (CH3, C-16´,16´´).
Vaccenic acid (5)
White amorphous powder, freely soluble in MeOH and insoluble in CH2Cl2, m.p 39-40 oC, ESIMS m/z 281.25 (100%) [M -H]- indicating a molecular formula C18H34O2.
1H NMR (CDCl3, 500 MHz):δH (ppm):5.35 (2H, m, J=4Hz, H-11,12), 2.22 (2H, t, J=8Hz, H-2), 1.62 (2H, t, J=8Hz,H-3), 1.34, (22H, br s, -CH2 of fatty acid), 2.05 (4H, m, J=4Hz, H-10,13), 1.34, (22H, br s), 1.40, (22H, br s), 0.92, (3H, t, J=8Hz, H-18 ), 13CNMR (CDCl3, 125 MHz, , assignment by APT and HMQC)δC (ppm):180 (C,C-1), 129.45 ,127.66 (CH,C-11,12), 36.39 (CH2, C-2), 26.7 (CH2, C-10,13), 25.82 (CH2, C-3), 31.67 (CH2 , C-16), 22.35 (CH2 ,C-17), 13.42 (CH3 , C-18), 28.98-29.48 (-CH2 of fatty acid)
1,3dicapryloyl 2- vaccenoyl-glycerol (6)
Yellowish white semisolid masses, it is freely soluble in CH2Cl2 and hexane, ESIMS m/z 685.6[M +Na]+ indicating a molecular formulaC41H76O6.1H NMR (CDCl3, 500 MHz)δH (ppm): 5.37 (2H, br, CH=CH), 5.29 (1H,m, -(C(O)O-CH2)2-CH-O-), 4.33 , 4.1 (1H each, dd-(C(O)O-CH2)2-CH-O-), 2.39 (2H, q, J=8 Hz , CH2C(O)O)2.04 (2H,m, CH2-C=), ), 1.64 (4H, CH2CH2C=O), 1.28 (48 H, (CH2)n, 0.95(9H, CH3 of fatty acid).13CNMR (CDCl3, 125 MHz) δC (ppm) : 173.3, 172.8 C(O)O, 130.0-129.8 CH=CH ,68, -(C(O)O-CH2)2-CH-O-, 62 -(C(O)O-CH2)2-CH-O-),34.1 CH2C(O)O,27.2 CH2-C=), 29.1-29.7 (CH2)n), , 24.9 CH2CH2C=O, 14.2 CH3.
Vaccenolymonoglyceride(7)
Yellowish white semisolid masses, itisfreely soluble in CH2Cl2 and hexane, ESIMS m/z 336 indicating a molecular formulaC21H40O4. 1H NMR (CDCl3, 500 MHz):δH (ppm)5.23 (2H, br, CH=CH), 4.07 (1H,m, -(C(O)O-CH2)2-CH-O-), 3.59 (1H, dd, J= 11.1, 5.9,-(C(O)O-CH2)2-CH-O-), 2.36 (2H, q, J=8 Hz , CH2C(O)O), 2.05 (2H,m, CH2-C=), 1.64 (4H, CH2CH2C=O), 1.28 (48 H, (CH2)n), 0.95(3H,t, CH3 of fattyacid) 13CNMR (CDCl3, 125 MHz)δC (ppm):172.5C(O)O, 68.3(-(C(O)O-CH2)2-CH-O-), 129.7-128.8 CH=CH, 62.4 -(C(O)O-CH2)2-CH-O-), 34.2 CH2C(O)O, 29.0-29.5(CH2)n),25.2 (CH2-C=), 25.6 (CH2CH2C=O), 14.1(CH3).
Results and discussion
Characterization of compounds
Compound 1
The NMR spectrum showed features which are characteristic for a typical withanolide steroidal skeleton with28 carbon signals of which two olefinic at δ 151.4 and 120.5, one carbonyl at δ 166.4, one oxygenated methine at δ 81.4and a methylene atδ 32.7representing a 6-membered lactone ring.Three olefinic double bonds resonating at δ 116.56, δ 141.45 and δ 125.59with their protons at δ 5.97, δ 6.99 and δ 5.79 were placed at C-2, C-3 and C-4 respectively.4 oxygenated carbons assigned as (C-14, C-17, C-20 andC-6)through HMBC correlations.
The structure of 1 was identified as Withaperuvin C and this conclusion was further supported by comparison of spectral data with previously published ones [11]. It is worth mentioning that, it is the first report on the isolation of withaperuvin C from W. Somnifera leaves.
Compound 2
The NMR spectrum had the general features of withanolides with great similarity with that of withanolide E but lacking any olefinic signals [5]. Instead, two signals were observed at δ 2.56 (H-2a, d, J=8Hz) and 2.75 (H-2b, dd, J= 8Hz,4Hz) that suggested the presence of 2,3dihydrowithanolide. The carbonyl signal at C-1 is shifted downfield compared to withanolide E indicating the presence of non conjugated ketone (δ 211). Two signals appeared at δ H 3.45 (H-3 resonating with a carbon atδ C 72.9, HMQC) &δ H3.83 (H-4,δ C70.8) corresponding to two hydroxyl bearing methine groups. An additional signal at 3.16 (H-6) indicated the presence of 5β, 6β -epoxy group[12].Anethoxy group was observed at δ 63.03,δ H 1.07 (3H, t)and 15.77 δ H3.39[12]located at C-3 as a result of the observed HMBC correlations of the 2 protons (OCH2-1´) to C-4 and from H-3 to CH3-2´. HMBC correlations was helpful to ascertain the presence of 5β, 6β -epoxy groupand the two oxymethine groups in ring A in positions 3 and 4 respectively.
Compound 2 was thus identified as Phyperunolide F; (2, 3-dihydro-4β hydroxy-3β ethoxywithanolide E) and this conclusion was further supported by comparison with previously published data [13].
As natural products containing an ethoxy group are extremely rare, Phyperunolide F was considered probably to be an artifact formed from 4β-hydroxywithanolide E that has been reported in trace amounts in W. somnifera[14]. Phyperunolide F is formed by a Michael-type addition of the extraction solvent ethanol to an enone system during the process of extraction and purification [14,15].
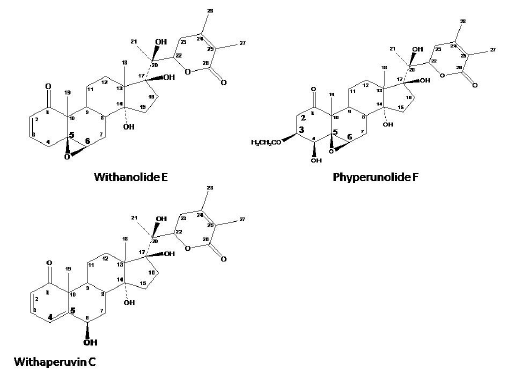
Fig. 1
Structures of withanolides
Compound 3
The IR analysis of compound 3showed the following signals: 3459 cm-1 (O-H stretching), 2919, 2851 (C-H2& C-H3 stretching), 1736 & 1064 (ester C=O & C-O stretching), 1215 & 1064 (S=O stretching), [16],794 and 720 (C-S stretching). ESIMS data showed the following peaks: m/z (% int.): 793.8 (100%) [M -H]- indicating a molecular formula C41H77O12SH. The 1H-NMR spectrum pointed to the presence of three isolated spin systems. The first one was assigned to glycerol moiety [δ 4.34 and 4.14, (2H, C-1), δ 5.13, (1H, C-2), δ 3.89 and 3.39, (2H, C-3)][17]. The second spin system signals were attributable to fatty acyl chain as the 1H-NMR spectrum showed signals of methylene groups at δ 1.24, 48H, The signal at δ 2.27 is characteristic of protons on carbon 2 of fatty acid, and the resonance at δ 1.5 is a feature of fatty acyl protons on the third carbon[18].The third spin system indicated the presence of a glycosyl moiety: one anomeric proton, a sequence of four oxymethynes and one deshielded methylene suggested the presence of hexapyranose structure.
COSY correlations were very important to confirm the spin systems; An ABMXY (H - H) coupling system[12]confirmed the glycerol moiety at δ4.34( (d, J 1a,1b= 12Hz, H-1a) and δ4.15 (q, J 1b,1a= 12Hz, J 1b,2= 5Hz, H-1b) [AB- part] , the second signal in glycerol was observed at δ 5.13 (d, J 2,1b= 5.3Hz, H-2)[M-part] and the third characteristic signals[XY- part] at δ 3.89 (q, J 3a,3b= 16Hz, J 3a,2= 2Hz, H-3a) and at δ 3.34(br s, H-3b)
The 13C-NMR spectrum of the compound confirmedthe features of a glucoglycerolipid spectrum; oxygenated carbons at (δ 63.1, 70.2 and 65.12) that were attributed to the glycerol carbons. Carbonyl carbon signals were observed at δ 172.98 and 172.82 indicating the presence of two acyl groups. Six oxygenated carbons signals; one for the anomeric carbon δ 98.3 (C-1´´´), four oxymethyne signals at δ 72.09, 73.3, 74.7 and 69.02 that were attributed to (C-2´´´, C-3´´´, C-4´´´ and C-5´´´) respectively of the sugar moiety. The carbon signal of C-6 was shifted upfield (δ 55), indicating a sulfated group rather than oxygen linkage at this position as a sulfodeoxyyhexozylunit [16] The chemical shifts of these carbon atoms suggested it to be a sulfoquinovopyranose unit.
Regarding the fatty acyl side chains; the presence of four methylene groups at δ 33.9, 34.04 and 24.93 represented C-3 and C-2 of the two fatty acyl side chains. Besides, the remaining methylene carbon shifts are clustered within asmall region of the spectrum at about δ 31.78- 22.57 (often referred to as the “methylene envelope”) [19]. The absence of olefinic carbon signals in 13C-NMR spectrum confirmed the nature of the side chain as saturated fatty acyl. The presence of 2 methyl singlets at δ 14.39 ascertained the presence of two fatty acyl side chains.
Assignments of all functional groups were achieved by 13C-APT, 1H-1H COSY, HSQC and HMBC spectra.
HMBC correlations from 2H-1 to C-1´ at δ 172.82 and the cross peak from H-2 to C-1´´ at δ 172.98 indicated the presence of (1, 2 diacyl glycerol moiety).
Confirmation of the linkage of the glycosyl moiety at position 3 of glycerol was based on the HMBC cross-peak between the anomeric proton H-1´´´ and C-3 (δ 65.12) of glycerol.
The anomeric proton also showed HBMC correlations with C-3´´´ and C-5´´´ of the sulfoquinovosylgluco moiety at δ 73.3 and 69.02. Also the correlations form H-6b´´´ to C-5´´´ (δ 69.02) and from H-6a´´´ to C-5´´´ and C-4´´´ confirmed the presence of a sulphonyl group attached at C-6´´´ carbon, of the sugar, instead of the hydroxyl group of glucose
All these findings strongly suggest that compound 4 is a sulfoglycolipid compound belonging to sulfoquinovosyldiacylglycerol (SQDG) class.
The mass spectrum supported the structure of the compound as it showed the distribution of isotopologues as 10:4:1.5 (for m/z 794, 795 and 796, respectively) [20]. The high intensity of the heavy isotopologues at m/z 794 is an indicative for the presence of a sulfur element in the molecules, occurring because of the presence of >4% of the heavy isotope (34S) in lipid fractions from vegetable sources. MS- MS spectrum was very informative to identify the acyl substituents at positions 1 and 2 in SQDG. The major product ion was found at m/z 555.4, representing the ketene (K) loss of a palmitic acid (16:0) molecule [M - H- C14H29CH = C = O]-. Besides, a relatively low intensity peak was detected at m/z 537.4, representing the neutral loss of a palmitic acid molecule [M - H- C15H31COOH]-. Other daughter ions that were detected at m/z 778 and 765 indicated the loss of -CH3 and -CH2CH3 groups from one of the palmitic ester groups [21].
These findings confirm that compound 3is a 16:0/16:0 SQDG and that the two fatty acyl moieties are identical and correspond to palmitic acid [21,22].
The structure of the compound 3was identified as 1,2-di-O-palmitoyl-3-O-(6´´´-sulfo-α-D-quinovopyranosyl)-glycerol. This conclusion was further supported by comparison with published data [16,23].
Previous studies have reported extensive biological effects for SQDG, such as DNA polymerase inhibition, immunosuppressive effect, antiviral activity mainly against the human immunodeficiency virus (HIV-1), antimicrobial and anti-tumor activities [21,24].
Similar products have been reported earlier from leaves of Byrsonima crassifolia , Azadirachta indica and Alfalfa leaves[25,26].In addition to many members of family Solanacae [27].Up to our knowledge, it is the first report on the isolation of SQDG compound from the leaves of W. Somnifera.
Compound 4
After examination of the NMR spectra, measuring melting point and comparing with published data the compound was identified as β-sitosterolglucoside [28].
Compound 5
The ESI-MS showed a peak at m/z 281.25 (100%) [M -H]- indicating a molecular formula C18H34O2with one site of unsaturation.
The 13C-APT spectrum (Table 3) showed 18 carbon signals of which two olefinic signals were detected at (δC 127.66, 129.45 and δH 5.35,2H, m). Together with a carbonyl signal at δC 180 indicated that 5 is a free monounsaturated fatty acid. Signal at (δC 13.42, δH 0.92, 3H, t) was attributed to the terminal methyl (CH3-18). The absence of allylic carbon signals at δ (32.5-32.7) confirmed that the double bond in the fatty acyl chain is cis rather than trans [19], the methylene hydrogensin β and α positions in relation to the carbonyl group have characteristic signals at δH 1.62 and 2.22 respectively.
HMBC and COSY correlations in addition to melting point determination 39-40 oCwere of great importance to distinguish between the four isomeric fatty acids with molecular formula C18H34O2 namely; [Oleic acid (18:1, ∆9 cis), Petroselinic acid (18:1, ∆6 cis), Vaccenic acid (18:1, ∆11 trans) and cis- Vaccenic acid (18:1, ∆11 cis)] previously isolated from plants [29]. Therefore, compound 5wasidentitifiedas (Z)-octadec-11-enoic acid commonly known as cis-11 Vaccenic acid [30]. The NMR and MS data were in agreement with those previously published [28,31]
Compound 6
ESI-MS data of compound 6 showed a peak atm/z: 685.6 [M+ Na]+ indicating a molecular weight 664 and molecular formula C41H76O6 in positive ion mode.IR bands at 3400,3100,2924,1745, 1635, 1167 cm-1 indicating the presence of hydroxyl, ester and olefinic groups
The 1H-NMR spectrum (Table 4) revealed the presence of two isolated spin systems. One of them with an AMX coupling systemδH 5.29, (1H, C-2) and [δH 4.15 and 4.34, (4H, C-1, 3)double doublets with a germinal coupling (J= 11.9 Hz) and a vicinal one (J= 6.0 or 4.2 Hz) with H-2[15]indicating a symmetrical triacylglycerol pattern[32]. Especially that the 13C-APT spectra showed only 2 signals at (δ 62.1 and 68.89 ppm). The other spin system signals were attributable to fatty acyl chain with signals similar to those of compound 5[33]. The presence of one unsaturated centre was deduced from the integration of the olefinic signal at δ5.38 (2H)their carbon atom resonate at δ 130.08 and 128.08. The integration of the CH3 signal at δ 0.9 (12H) indicated the presence of three methyl signal [34,35]
Carbonyl carbon signals arising from acyl groups were observed at δ 173.3 and 173.0. COSY, HSQC and HMBC correlations from H-2´, H-3´ and from the glycerol protons [(H-1a, 3a) & (H-1b, 3b)] to the carbonyl carbons at δ 173.3
ESI-MS showed certain characteristic peaks 491 (M-RCOO), 369 (M-RCOOCH2), 339 (RCO +74) for unsaturated C18 acid, 283 (RCO +128) for C10 acid [32, 36,37].
Alkaline hydrolsis was carried out and the isolated fatty acids identities were confirmed to be vaccenic and capric acids via comparing their NMR spectra and melting points. Therefore compound 6 identity was confirmed to be 1,3 dicaproyl,2-vaccenoyl-glycerol.
Compound 7
ESI-MS data of compound7showed a peak at m/z: 356.32 M+ indicating a molecular formula C21H40O4. The 1H-NMR spectrum revealed the presence of two isolated spin systems. One of them is for 1-monoacyl glycerol. The signal at 3.59 (dd, J = 11.11 and 5.99 Hz) belongs to the sn-1 protons of glycerol in 1-monoacyl glycerol and the signal at 4.07 is attributed to CH of glycerol and 5.23 is for C-18 monounsaturated fatty acid [37].
Alkaline hydrolsis was carried out and the isolated fatty acid identity was confirmed to be vaccenic acid via comparing its NMR spectra and melting points. Therefore, compound 7identity was confirmed to be vaccenolymonoglyceride.
TLC screening of the aqueous fraction of the ripe fruit extractrevealed the presence of amino acids as its major components. Two of them were identified according to their Rf values and color after spraying with ninhydrin on TLC as L-Dopa andL-Asparagine.
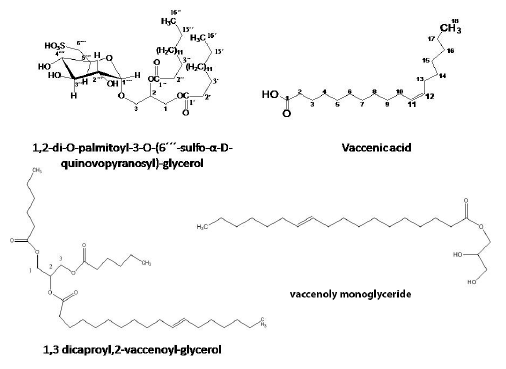
Fig. 2
Structures of compounds 4-7
Anticholinesterase activity
Comparing the activity of the different successive fractions of W. Somnifera ripe fruits, The aqueous and light petroleum fraction were the most potent AChE inhibitors with the lowest IC50 value of 0.55 and 0.56 mg/ml respectively, followed by fraction “WR” that has IC50 value of 0.64 mg/ml then methylene chloride and ethyl acetate fractions with IC50 1.45 and 1.07mg/ml compared to physostigmine which showed IC50 of 2.8×10-4mg/ml.
Conclusion
This study confirmed the activity of the ripe fruits extract of Egyptian Withania somniferaagainst Alzheimer's disease relating the activity to the phytoconstituents present in the aqueous and light petroleum fractions of the fruits. Previous studies on the fruit reported that withanamides are the metabolites responsible for such biological activity. In the present study, we have shown that other lipids contribute to this activity. The current study also reports the isolation of SQDG, withaperuvin C and phyperunolide, vaccenic acid for the first time from W. Somnifera. In addition to the novel compounds 1,3 dicaproyl,2-vaccenoyl-glyceroland vaccenolymonoglyceride.
Acknowledgment
The authors would like to thank prof. Dr. Doaa Ghareeb, Department of Biochemistry, Faculty of science, Alexandria university, Egypt for providing the necessary equipment and chemicals to carry out the anticholinesterase activity. Authors are also grateful to Dr. Armelle Vallat, Institute of Chemistry, University of Neuchâtel, Switzerland for carrying out the mass spectral analysis.
References
1. Sehgal N, Gupta A,Valli RK, Joshi SD, Mills JT,Hamel E, et al. PNAS USA.; 2012, 109(9), 3510-3515.
2. Dar NJ, Hamid A, Ahmad M.CMLS. 2015, 72(23), 4445-4460.
3. Kumar V, Dey A, Hadimani M, Marcović T, Emerald M. tang humanitas Med . 2015, 5, 1-13.
4. Mishra LC, Singh BB, Dagenais S. Altern med rev. 2000, 5(4), 334-346.
5. Nassra R., Rahma Sr. Mahrous R, Fathy H,.Abu El-Khair R and Omar A. EJMP. 2017, 21 (3), 1-12. https://doi.org/10.9734/EJMP/2017/38239
6. Glotter E, Kirson I, Abraham A, Lavie D.Tetrahedron. 1973, 29(10), 1353-1364.
7. Kirson I, GlotterE;Lavie D, Abraham A. J Chem Soc C, 1971, 2032-2044.
8. Mahrous R, Ghareeb D, Fathy H, EL-Khair R, Abdallah A. Med Aromat Plants 2017, 6:285. https://doi.org/10.4172/2167-0412.1000285
9. Mahrous RS, Fathy HM, Abu EL-Khair RM, Omar AA. . J Sep Sci. 2017, 1-7. https://doi.org/10.1002/jssc.201700764
10. Ellman G, Courtney K, Andres V, Jr., Feather-Stone R. Biochem pharmacol. 1961, 7:88-95. https://doi.org/10.1016/0006-2952(61)90145-9
11. Damu A., Kuo P., Su C., Kuo T., Chen T., Bastow K. etal. J nat prod. 2007, 70 (7):1146-1152. https://doi.org/10.1021/np0701374
12. Silverstein R;Webster F.;Kiemle D;Bryce D.Spectrometric Identification of Organic Compounds: Wiley.2014.
13. Lan Y-H, Chang FR, Pan J, Wu C, Wu S, Chen S, etal. Food chem. 2009,116 (2):462-469.https://doi.org/10.1016/j.foodchem.2009.02.061
14. Chen LX, He H, Qiu F. Nat Prod Rep, 2011, 28(4): 705-740. https://doi.org/10.1039/c0np00045k
15. Cao CM, Zhang H, Gallagher RJ, Timmermann BN. J Nat Prod. 2013,76 (11): 161-164. https://doi.org/10.1021/np400296s
16. Wang H;Li Y-L;Shen W-Z;Rui W;Ma X-J;Cen Y-Z. Bot Mar. 2007, 50(3):185-90. https://doi.org/10.1515/BOT.2007.022
17. Jung JH;Lee H;Kang SS. Phytochem.1996, 42(2):447-52. https://doi.org/10.1016/0031-9422(95)00929-9
18. Coleman J. J Biol Chem. 1990, Oct 05;265(28):17215-21.
19. Lie Ken Jie MSF;Mustafa J. Lipids. 1997, ;32(10):1019-34.
20. De Souza L, Sassaki G., Romanos M., Barreto-Bergter E. Mar Drugs. 2012, Apr; 10(4): 918-931.
21. Zianni R, Bianco G., Lelario F, Losito I, Palmisano F, Cataldi T. JMS. 2013; 48 (2): 205-2015.
22. Al-Fadhli A;Wahidulla S;D'Souza L.. Glycobiology. 2006, Oct;16(10):902-15.
23. Cantillo-Ciau Z;Moo-Puc R;Quijano L;Freile-Pelegrín Y. Mar Drugs. 2010, 8 p. 1292-304.
24. Sassaki GL; Gorin PA; Tischer CA; Iacomini M. Glycobiology. 2001, Apr;11(4):345-51.
25. Rastrelli L;De Tommasi N;Berger I;Caceres A;Saravia A;De Simone F. Phytochem. 1997, 45 (4), 647-650.
26. Chatterjee R;Singh O;Pachuau L;Malik SP;Paul M;Bhadra K, et al. Bioorg Med Chem lett. 2010, Nov 15;20(22):6699-702.
27. Kenrick JR;Bishop DG. Plant Physiol. 1986, Aug;81(4):946-9.
28. Khan N.andHossain M.. J Pharmacogn Phytochem 2015, 4:5-7.
29. Avato P, Fannizzi P, Rosito I. Lipids. 2001;36 (8):845-850.
30. Vlahov G, Chepkwony P, Ndalut P. J Agric Food Chem. 2002, 50 (5): 970-975.
31. Kamalud A., Bumpu F, Strong F. J Am Chem. Soc. 1948, 70: 3391-3394.
32. Gusntone FD. Chem Phys Lipids. 1991, 58 (3): 219-224.
33. Duffy P., Quinn S., Roche H, Evans P. Tetrahedron. 2006, 62 (20):438-4843.
34. Guillen M, Ruiz F. Eur j Lipid Sci Technol. 2003, 105 (9):502-507.
35. Sacchi R, Addeo F, Paolillo L. Magn Reson Chem. 1997, 35 (13).
36. McAnoy A., Wurobert, Murphy C. JASMS. 2005, 16 (9) 1498-1509.
37. Kalo, P. and Kemppinen, A. J Am Oil Chem Soc 1993, 70: 1209.
Author notes
*Corresponding author: Hoda M. Fathy, e-mail: hodasherif@hotmail.com, hoda.sherif@alexu.edu.eg, Phone:00201001223411
