Journal of the Mexican Chemical Society, vol. 63, no. 03, 2019
Sociedad Química de México A.C.
Daniel J. Freppon
Iowa State University, USA
Ames Laboratory, Ames, Iowa 50011., USA
Long Men
Iowa State University, USA
Ames Laboratory, Ames, Iowa 50011., USA
Ujjal Bhattacharjee
Iowa State University, USA
Ames Laboratory, Ames, Iowa 50011., USA
Bryan A. Rosales
Iowa State University, USA
Feng Zhu
Iowa State University, USA
Jacob W. Petrich
Iowa State University, USA
Ames Laboratory, Ames, Iowa 50011., USA
Date received: 07 July 2018
Date accepted: 09 April 2019
Funding
Funding source: Iowa State University
Contract number: DE-AC02-07CH11358
Abstract: An optimized synthetic procedure for preparing photostable nanocrystalline methylammonium lead halide materials is reported. The procedure was developed by adjusting the lead halide to methylammonium/octylammonium halide precursor ratio. At a high precursor ratio (1:3), a blue-shifted photoinduced luminescence peak is measured at 642 nm for CH3NH3PbI3 with 0.01 to 12 mJ pulsed-laser irradiation. The appearance of this peak is reversible over 300 min upon blocking the irradiation. In order to determine if the peak is the result of a phase change, in situ x-ray diffraction measurements were performed. No phase change was measured with an irradiance that causes the appearance of the photoinduced luminescence peak. Luminescence microscpectroscopy measurements showed that the use of a lower precursor ratio (1:1.5) produces CH3NH3PbI3 and CH3NH3PbBr3 perovskites that are stable over 4 min of illumination. Given the lack of a measured phase change, and the dependence on the precursor ratio, the photoinduced luminesce peak may derive from surface trap states. The enhanced photostability of the resulting perovskite nanocrystals produced with the optimized synthetic procedure supports their use in stable optoelectronic devices.
Keywords: surface traps, nanocrystalline perovskites, photostability, single nanocrystal analysis, synthetic optimization, optoelectronics.
Resumen: Se reporta un proceso sintético optimizado para preparar materiales haluros de metilamonioplomo nanocristalinos fotoestables. El proceso fue desarrollado ajustando la relación entre los precursores de haluro de metilamonio y haluro de octilamonio. Con una relación de precursores alta (1:3), un pico luminiscente desviado al azul a 642 nm fue detectado para CH3NH3PbI3 con irradiación de láser de pulso entre 0.01 y 12 mJ. La aparición de éste pico es reversible hasta por 300 min después de cesar la irradiación. Para determinar si éste pico se debe a una transición de fase, se hicieron mediciones de difracción de rayos-X in situ. Ningún cambio de fase fue detectado con niveles de irradiación a los cuales se observa el pico luminiscente. Mediciones luminiscentes macroespectroscópicas muestran que con una relación de precursores más baja (1:1.5) se producen perovskitas CH3NH3PbI3 y CH3NH3PbBr3 que son estables hasta con 4 min de iluminación. Dado que no se observa un cambio de fase, y la dependencia sobre la relación de precursores, el pico luminiscente generado bajo iluminación podría derivarse de estados trampa de superficie. La fotoestabilidad incrementada de los nanocristales de perovsita obtenidos por el procedimiento sintético optimizado ayudan a su aplicación en dispositivos optoelectrónicos más estables.
Palabras clave: trampas de superficie, perovskitas nanocristalinas, fotoestabilidad, análisis de nanocristal individual, optimización sintética, optoelectrónica.
Introduction
Organolead halide perovskite semiconductors of general composition RPbX3 (R = organic monocation, such as CH3NH3 +; X = halide, such as I- or Br-) have drawn attention as both photovoltaic [1, 2] and optoelectronic [3] materials. Broad light absorption and long carrier diffusion lengths make perovskites ideal light harvesters [4]. The certified power conversion efficiency of perovskite solar cells surged from 3.8% to over 22% in the last 8 years [5-10]. In spite of these many advantages, organometal halide perovskites suffer from instability against moisture, heat and light [11-13]. A deeper understanding of the fundamental physical and chemical behavior of perovskites could help in mitigating these instability issues, thus enabling their implementation and deployment into many energy technologies [14, 15].
Efforts to improve the physical and chemical properties of perovskites have focused primarily on tuning their composition or dimensionality [16,17]. Compositional and dimensional control are useful in tuning the bandgap energies of some perovskite materials [18,19]. Partial substitution with long alkylammonium cations leads to low dimensional perovskites [20-22], some of which exhibit enhanced moisture stability. Control of optoelectronic properties through mixing cations and halides has been widely exploited in enhancing the power conversion efficiency of perovskite solar cells [14,23-25]. Compositional variants of halide perovskites have bandgap and luminescence energies that cover the entire visible spectrum [9, 26-28]. Halide substitution also leads to enhanced stability, as CH3NH3PbI3 solar cells doped with Br show long-lasting resistance against humidity [29, 30]. CH3NH3PbBr3 displays lower sensitivity to concentrated sunlight compared to CH3NH3PbI3 [31].
A few reports describe the unusual photophysical behavior of organometal halide perovskites, specifically a reversible shift in photoluminescence maximum (λmax) under thermal and photochemical conditions [32-34] Gottesman et al. observed a decrease in luminescence intensity and increase in the 108 cm-1 Raman band of CH3NH3PbI3 solar cells, and attributed these changes to slow photoinduced structural changes since the timescale of the change was not consistent with an electronic process [34]. Sadhanala and co-workers measured two absorption peaks in freshly-prepared, mixed-halide CH3NH3Pb(Br1-xIx)3 perovskite films. A single absorption band, however, was measured after aging the film for 21 days [33]. Hoke et al. have also reported light-induced changes to the absorption and photoluminescence spectra of CH3NH3Pb(Br1-xIx)3 perovskite films. They attribute these behaviors to reversible crystalline changes and trap states [32]. Structural defects and ion migration contribute to the notorious photocurrent hysteresis [35, 36] and specific quantum efficiency [37] that characterizes perovskite semiconductors and devices. Unusual photophysics that are caused by surface defects may become even more prominent in nanocrystalline perovskites [38].
Trap states in perovskites. CH3NH3PbI3 has a broad emission band and a high quantum yield of photoluminescence. The nature of the radiative decay channels and the spectral broadening mechanisms are largely determined by phonon coupling effects and defects or trap states [39]. Trap states are likely to occur at the surface of perovskite nanocrystals [38]. Vacancies, such as ionic defects like Pb+, I-, and CH3NH3 + , can also form shallow trap states and reduce carrier lifetimes [40]. Strong covalency of the Pb cations and I anions lead to the formation of Pb dimers and iodide trimers, which are responsible for transition levels that can serve as recombination centers that reduce solar-cell performance [41]. Trap states increase the frequency of non-radiative recombination, which reduces the quantum yield [42]. Density-functional theory studies have revealed the unusual defect physics of CH3NH3PbX3 [41, 43-46]. For example, shallow point defects could explain in part the ultra-high open-circuit voltages of CH3NH3PbBr3 solar cells [46].
Synthetic conditions play an important role in the formation of trap states. For example, perovskites grown under high iodine concentrations are likely to have defects (lead atom substituted by iodide) and a high density of deep electronic traps (recombination centers) that cause short diffusion lengths and poor photovoltaic performance [47]. Supramolecular halogen bond complexation can passivate the under-coordinated iodine ions, which can reduce trap sites near the perovskite surface [48]. Lewis bases are used to passivate under-coordinated lead atoms, and treated perovskites exhibited reduced electron-hole recombination and consequently longer photoluminescence lifetimes [49]. Adding fullerene layers has also proven to be an effective way to passivate the charge trap states and get rid of photocurrent hysteresis [35, 50].
Building on our previous work [22], a systematic synthesis of photostable nanocrystalline CH3NH3PbX3 is demonstrated. Optimization of the CH3NH3PbI3 alkyl halide to lead halide precursor ratio, inhibited the appearance of a reversible photoinduced 630-nm photoluminescence peak that may be derived from surface traps. Using an optimized synthetic procedure, luminescence of single CH3NH3PbI3 and CH3NH3PbBr3 nanocrystals was stable over 240 seconds with over 1 × 105 W/cm2 irradiance. The reported optimized synthesis may be applicable to other organometal halide perovskites, for example those with mixed halide composition.
Experimental
Materials
Lead(II) bromide (≥ 98%), N,N-dimethylformamide (DMF), lead(II) iodide (99%), methylamine (33 wt% in ethanol), n-octylamine (99%), and toluene (anhydrous, 99.8%) were purchased from Sigma-Aldrich. Hydrobromic acid (ACS, 47.0-49.0%), hydroiodic acid (ACS, 55-58%), and oleic acid (tech., 90%) from Alfa-Aesar; diethyl ether from Baker; toluene (99.9%) and acetonitrile (99.9%) from Fisher. Materials were used as received unless specified otherwise.
Synthesis
Ammonium Halides. Dimensionality control is achieved using a bulky alkylammonium cation as a capping ligand. Hydrogen halides were prepared by a modified literature procedure [45]. Briefly, hydroiodic acid (10 mL, 0.075 mol) or hydrobromic acid (8.6 mL, 0.075 mol) was added to a solution of excess methylamine (24 mL, 0.192 mol) in ethanol (100 mL) at 0 °C, and the mixture stirred at this temperature for 2 h. The sample was concentrated under vacuum, and the resulting powder dried under dynamic vacuum at 60 °C for 12 h and recrystallized from ethanol. Both n-octylammonium iodide (CH3(CH2)7NH3I) and n-octylammonium bromide (CH3(CH2)7NH3Br) were washed repeatedly with diethyl ether and dried under dynamic vacuum before use.
Nanocrystalline CH3NH3PbX3. PbX2 (0.008 mmol), CH3NH3X (0.012 mmol) and CH3(CH2)7NH3X (0.012 mmol) were dissolved in a mixture of acetonitrile (20 mL) and DMF (200 µL). 4 mL of the resulting precursor solution was rapidly injected into toluene (15 mL) while stirring. After 24 h stirring at 20 °C, solids were isolated by centrifugation (10 min at 4000 rpm) and purified by washing with toluene (5 mL) followed by re-centrifugation.
Structural Characterization
X-ray diffraction (XRD) was collected on a Rigaku Ultima IV (40 kV, 44 mA). A Cu KR source was used for radiation. A quartz sample holder was used as a substrate for drop-casted toluene solvated samples. Details of the in situ measurements were as previously reported [51]. Transmission Electron Microscopy (TEM) images were collected using a FEI Tecnai G2 F20 field-emission TEM capable of 200 kV with a point-to-point resolution of less than 0.25 nm having a 0.10-nm line-to-line resolution. Dilute solutions were prepared in toluene and 2 to 3 drops of each product were placed onto carbon-coated copper grids. TEM images were used to measure the particle dimensions and were processed using the ImageJ program. Typically, more than 100 particles were counted in each case. Uncertainties in all measurements are reported as standard deviations.
Optical Characterization
Solution-phase optical extinction (absorption plus scattering) spectra were collected using an Agilent 8453 UV/Vis spectrophotometer equipped with a photodiode array. Solvent absorption was subtracted from all spectra. Drop-casted solid films of each sample were measured using diffuse reflectance (SL1 Tungsten Halogen lamp (vis-IR), a SL3 Deuterium Lamp (UV), and a BLACK-Comet C-SR-100 Spectrometer). A Horiba-Jobin Yvon Nanolog scanning spectrofluorometer equipped with a photomultiplier detector was used to collect steady-state PL spectra. Quantum yields for each nanocrystalline perovskite were calculated by comparing luminescence intensities of Rhodamine 590 or 640 [52].
For the luminescence microspectroscopy of single nanocrystals, a DM IRBE microscope (Leica, Wetzlar, Germany) was employed. Monochromatic illumination with a 532 nm laser (Coherent, Santa Clara, CA) was used to excite CH3NH3PbI3, and a 488 nm Argon ion laser (Uniphase, San Jose, CA) for CH3NH3PbBr3. An oil-immersion objetctive producing a spot with a 0.28 ± 0.03 μm diameter provided an excitation power density of 1.6 × 105 W∙cm−2. PL was collected using a HoloSpec f/1.8i spectrograph (Kaiser Optical Systems, Ann Arbor, MI, USA) equipped with a broad-range grating (HFG-650, Kaiser Optical Systems) and a charged-coupled device (CCD) (Newton 940, Andor Technology, Belfast, UK) with a collection binning time of 0.05 s. 2400 spectra were collected every 0.09873 seconds.
Results and discussion
General Synthesis and Characterization
Nanocrystalline organolead halide perovskites are prepared by dissolving (a) PbX2, (b) CH3NH3X, and (c) CH3(CH2)7NH3X (X = I or Br) precursors in a polar solvent such as dimethyl formamide (DMF, ε = 38.25) or acetonitrile (ε = 36.64), followed by quick injection into a less polar solvent such as toluene (ε = 2.379) as shown in Scheme 1 [22]. DMF, acetonitrile, or both are used because they provide excellent precursor solubility, as we reported previously for the synthesis of perovskite nanowires [22]. Introduction of a large ionic ligand, such as an ammonium or carboxylate-containing surfactant, decreases the perovskite particle size. In this study, the concentration of the CH3NH3X and larger alkyl ammonium halide, n-CH3(CH2)7NH3X, are equivalent. In this way low-dimensional nanocrystalline perovskites are synthesized [16, 38, 53]. Perovskite nanocrystals produced by this method contain methyl ammonium cations within their core, and octyl ammonium groups on their surface [54].
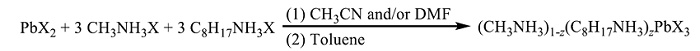
Scheme 1
Synthesis of nanocrystalline organolead halide (X = Br or I) perovskites prepared using a molar concentration of alkylammonium halide precursor that is 3× higher than that of the lead halide precursor; 0 < z << 1.
During the synthesis of iodide perovskites, the PbI2 precursor fails to dissolve completely in the co-solvent unless an excess of ammonium halide precursors is present. A very large excess of ammonium halides, however, irreversibly affects the optical properties of the resulting perovskites, as discussed below. Precursor reactivities and ease of forming solid solutions also affect the product. CH3NH3X salts dissociate differently in DMF depending on the specific halide (Scheme 2). For iodide, the preferred products are CH3NH3 + and I-, the conjugate base of HI, which is a strong acid in DMF and leads to a large conductivity (Scheme 2, Fig. 1 when [PbX2]0 = 0). For bromide the preferred products are CH3NH2 along with HBr, which is a weaker acid in DMF and leads to a smaller conductivity (Scheme 2, Fig. 1 when [PbX2]0 = 0). Thus, CH3NH3I is expected to be the most reactive ammonium halide precursor in DMF, generating free and readily available I- needed for perovskite formation that should be easily precipitated upon addition of a nonpolar solvent such as toluene.
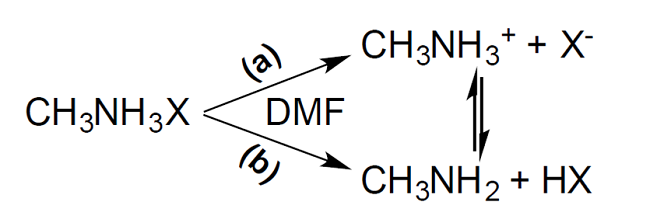
Scheme 2
Pathways of CH3NH3X dissociation in DMF.
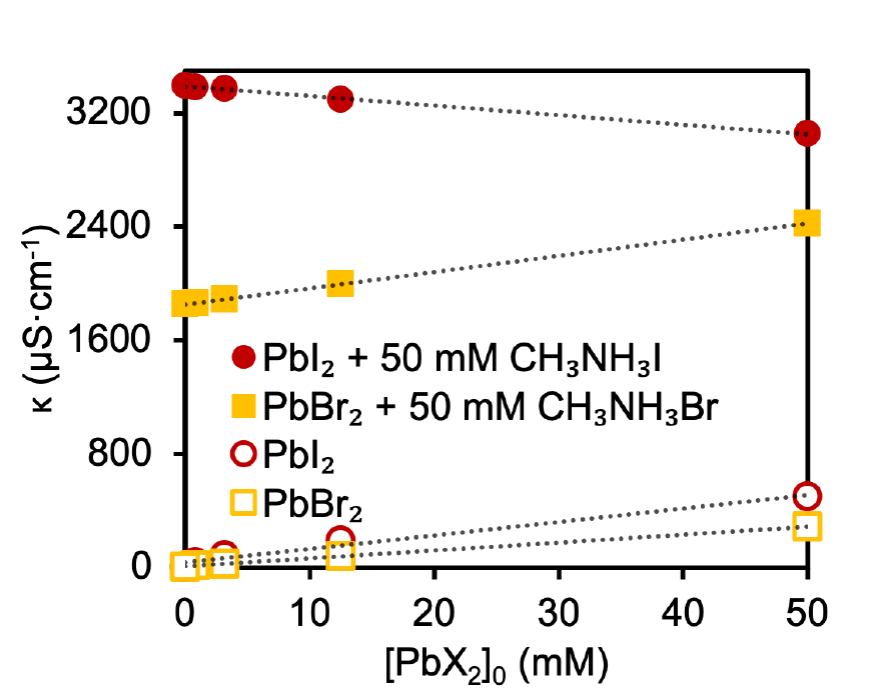
Fig. 1
Specific conductivity (κ) vs. PbX2 concentration with (solid) or without (hollow) 50 mM CH3NH3X.
To probe PbX2 precursor reactivity, we measured their specific conductivities (κ) in DMF with both the presence and absence of a set amount of the corresponding CH3NH3X (Fig. 1). As expected, in pure DMF, the conductivity increases linearly with PbX2 concentration. Conductivity is proportional to the number of ions in solution, thus enabling a comparison to the relative degree of dissociation and association. A steeper increase for the iodide case suggests that dissociation is slightly higher for PbI2 than for PbBr2. With the presence of 50 mM CH3NH3X in DMF, the conductivity actually decreases upon addition of PbI2 and slowly increases upon addition of PbBr2. When a 1 to 3 ratio of PbX2 to CH3NH3X is used, as is the case in the Scheme 1 perovskites, the conductivity is higher for PbI2/CH3NH3I than PbBr2/CH3NH3Br suggesting that Pb-Br binding in solution is stronger than Pb-I binding. A similar observation was reported by luminescence and transient absorption measurements [55].
Structural Analysis
Powder X-ray diffraction (XRD) patterns show the diffraction peaks corresponding to the <110> and <001> facets of the CH3NH3PbI3 and CH3NH3PbBr3 nanocrystals (Fig. 2). CH3NH3PbBr3 shifts to higher 2( values since bromine has a higher electronegativity compared to iodine, indicating some degree of solid solution, consistent with literature [56, 57]. Table 1 presents the lattice parameter of cubic CH3NH3PbBr3 and tetragonal CH3NH3PbI3 nanocrystalline perovskites as determined experimentally from XRD analysis.
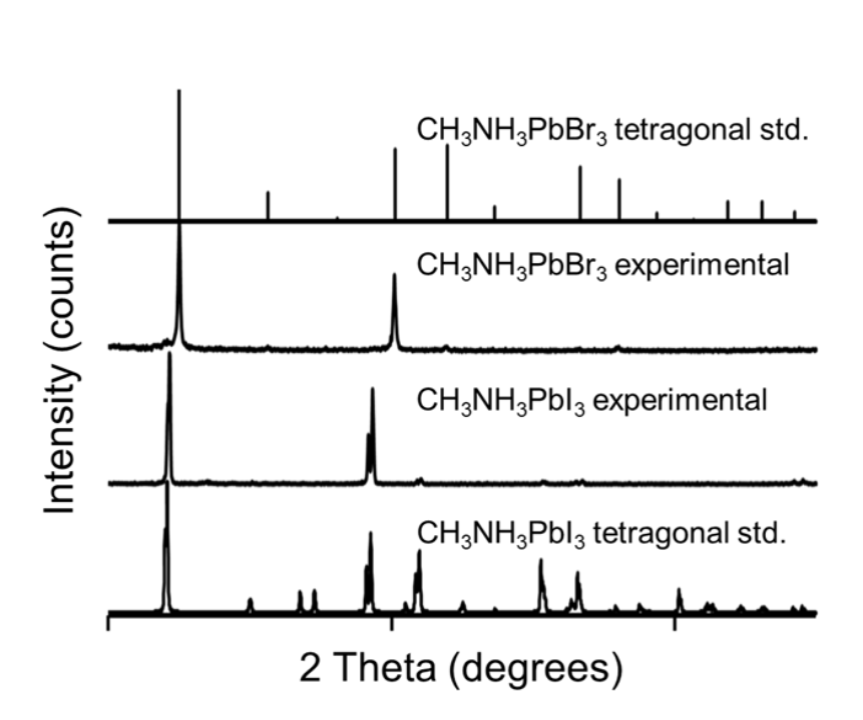
Fig. 2
Powder XRD of nanocrystalline organometal halide perovskites with corresponding standard (std.) diffraction patterns.
Characterization parameters of low-dimensional organometal halide perovskites.
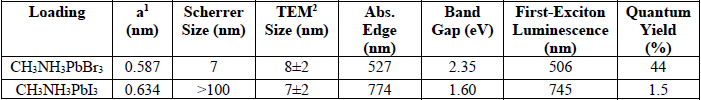
The transmission electron microscopy (TEM) images shown in Fig. 3 reveal that the perovskites are primarily nanospheres with an average size of 7 ± 2 nm for CH3NH3PbI3 and 8 ± 2 nm for CH3NH3PbBr3. The Bohr radii for bromide and iodide perovskites are 2 and 2.2 nm, respectively [58]. In addition to the nanospheres, the CH3NH3PbBr3 perovskites exhibit nanosheets making up ca. 10-20% of the total particles. As we previously reported, perovskite nanosheets are unstable under the TEM electron beam [22]. The presence of nanosheets is a contributing factor for the preferred orientation behavior observed by powder XRD for CH3NH3PbI3 perovskite nanocrystals.

Fig. 3
Representative TEM images of CH3NH3PbBr3 and CH 3NH3PbI3 nanocrystalline perovskites.
Optical Properties and Photostability
Using a spectrofluorometer with arc lamp illumination, the (λmax for CH3NH3PbI3 was determined to be 760 nm; and that for CH3NH3PbBr3, 518 nm. This is consistent with literature reports for perovskites of similar composition [59-61]. The CH3NH3PbBr3 perovskite sample has a 29-fold higher photoluminescence quantum yield (44%) compared to CH3NH3PbI3 (Table 1).
Nanocrystalline perovskites prepared using Scheme 1 with an a:b:c precursor ratio of 1:3:3 exhibited two luminescence peaks in a flash photolysis experiment with nanosecond pulsed Nd:YAG laser illumination. One peak had a (λmax at 745 nm (Eg) and the second peak had a (λmax at 642 nm (E>Eg) for CH3NH3PbI3 and 0.01 mJ pulsed-laser illumination (Fig. 4a). With increasing excitation energy the relative intensity of the 642 nm peak increases nonlinearly (Fig. 4b) and both peaks blue shift. With 12 mJ illumination, the (λmax are 626 nm and 725 nm.
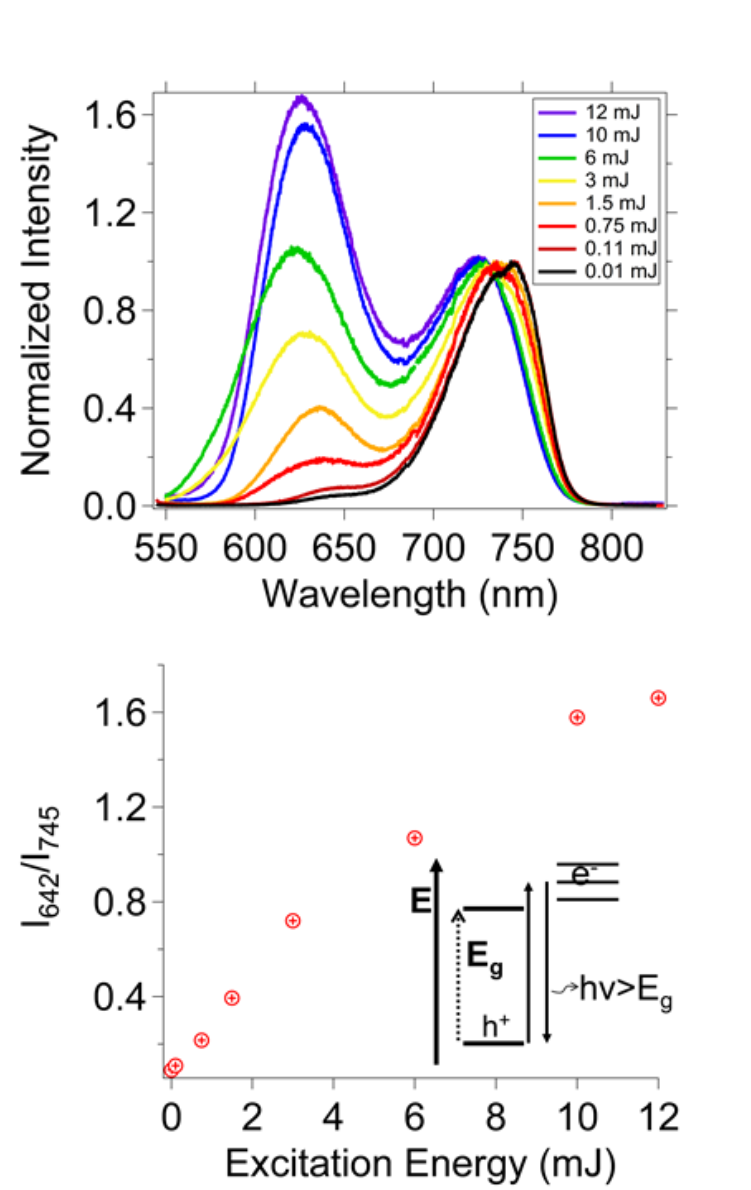
Fig. 4
(Top) Flash photolysis luminescence spectra of nanocrystalline CH3NH3PbI3, prepared using Scheme 1, as a function of excitation power. The spectra are normalized to the intensity at ~745 nm (at the λmax). (Bottom) Intensity ratios of the ~642nm (I642) and ~745 nm (I745) bands vs. incident laser power. The inset explains the band gap (Eg) of the nanocrystalline perovskite “vs” the higher energy trap state luminescence.
To determine if the photoinitiated increase in the 642 nm peak intensity was reversible, the luminescence spectrum of the CH3NH3PbI3 nanocrystals was monitored in a new-batch of nanocrystals after blocking the laser irradiation. After 300 minutes in the dark, the intensity of the band at 642 nm decreased due to a thermal relaxation mechanism. The photoinitiated reversible spectral changes may be the result of changes in the crystal phase or the population of surface traps.
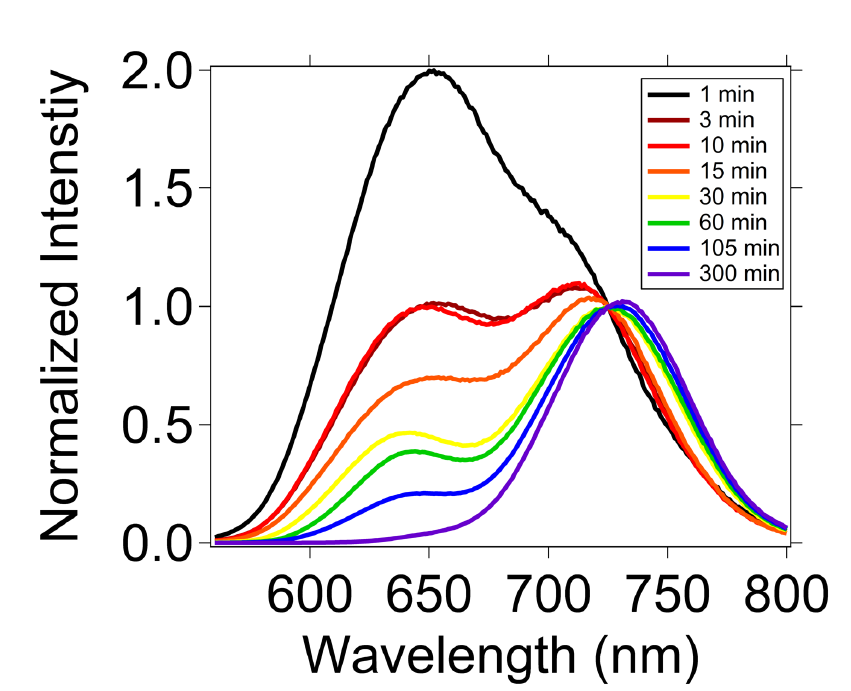
Fig. 5
Flash photolysis luminescence spectra of nanocrystalline CH3NH3PbI3 made with Scheme 1 showing thermal relaxation subsequent to laser illumination from a 532-nm, 15-mJ Nd:YAG laser providing nanosecond pulses. Time zero corresponds to the time when the laser was blocked from illuminating the sample.
In order to rule out phase changes as the cause of the photoinduced spectral changes, we conducted in situ XRD experiments to search for possible structural changes in the nanocrystalline perovskites during illumination with a pulsed laser. An open window was used to irradiate the samples while they were mounted in the XRD instrument as we have previously reported for other in situ XRD measurements [51]. The nanocrystalline material was in the solid state so no solvent effects were present. The CH3NH3PbI3 nanocrystalline perovskites exhibited no change in 2θ diffraction peak location or new peaks as a result of irradiation with a laser energy up to 15 mJ (Fig. 6). The in situ XRD analysis revealed no additional or shifted diffraction peaks, which indicates that a phase transition does not occur upon illumination.
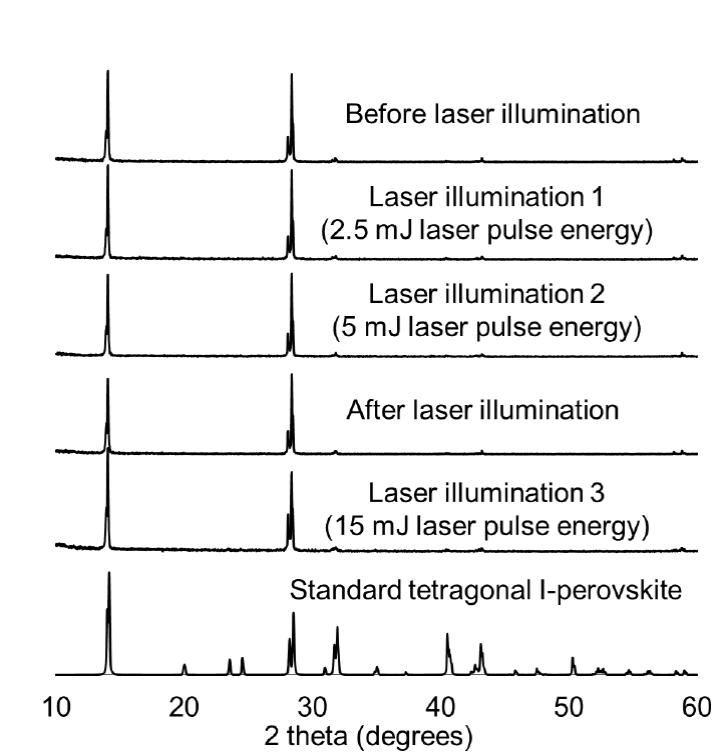
Fig. 6
X-ray diffraction patterns from nanocrystalline CH3NH3PbI3 perovskites at different pulse energies. The laser illuminated the sample during the entire time XRD data were collected.
Since a phase change was not measured upon irradiating the sample, the photoinitiated blue photoluminescence peak may result from surface trap states that are populated upon irradiation. In this case, the perovskites have traps with energies above the conduction band that emit at higher energies than the band gap (Fig. 4binset). An increasing population in trap states with increasing irradiation power could explain the increasing intensity of the 630-nm peak in going from 0.01 mJ to 12 mJ (Fig. 4). Excess precursor may lead to higher energy emissive states which differ from the traditional nanocrystalline lattice expected for CH3NH3PbI3 nanocrystals. By using a precursor ratio of 1:1.5:1.5 (Scheme 2), the CH3NH3PbBr3 and CH3NH3PbI3 exhibited only one luminescence peak at 540 nm and 800 nm, respectively (Fig. 7a, Fig. 8a). The disappearance of a second luminescence peak illustrates that bromide and iodide nanocrystalline perovskites prepared using Scheme 2 each have a single emitting state compared to the multiple emitting states observed for the nanocrystals prepared using Scheme 1 with a higher precursor ratio. This observation supports the hypothesis that a synthesis using the smaller ratio of precursors may result in nanocrystals with higher crystalline order and fewer defects compared to a synthesis using a larger precursor ratio.
Scheme 2
Optimized synthesis of nanocrystalline organolead halide (X = I or Br) perovskites prepared using a molar concentration of alkylammonium halide precursor that is 1.5× that of the lead halide precursor; 0 < z << 1.
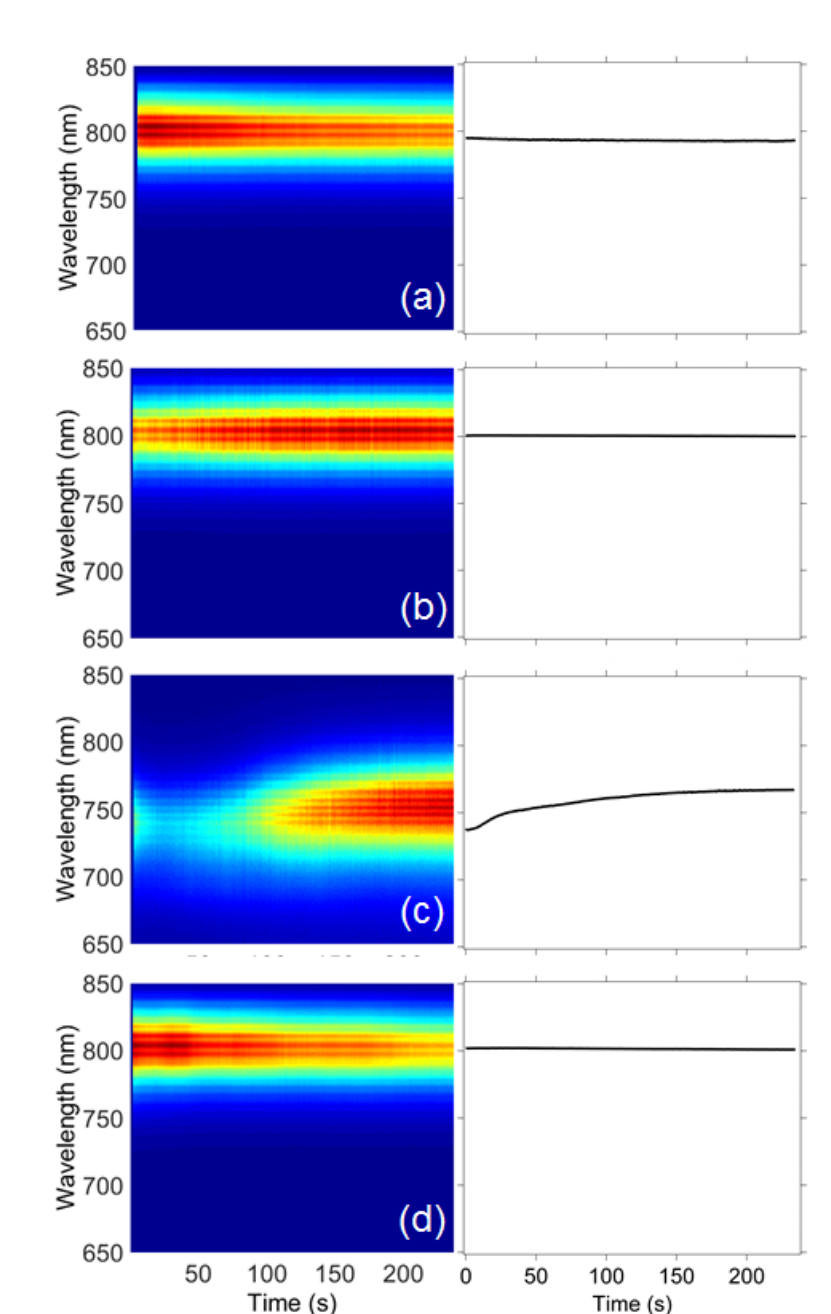
Fig. 7
Time-correlated luminescence microspectroscopy spectra of single CH3NH3PbI3 perovskites. The left column shows the plots of luminescence versus illumination time with a 532 nm laser (1.58 × 105 W/cm2) for CH3NH3PbI3 perovskites synthesized using Scheme 2. The samples are: (a) unwashed sample, (b) washed sample, (c) unwashed with excess precursor, and (d) washed with excess precursor sample. The right column shows the average λmax versus illumination time (n=3).
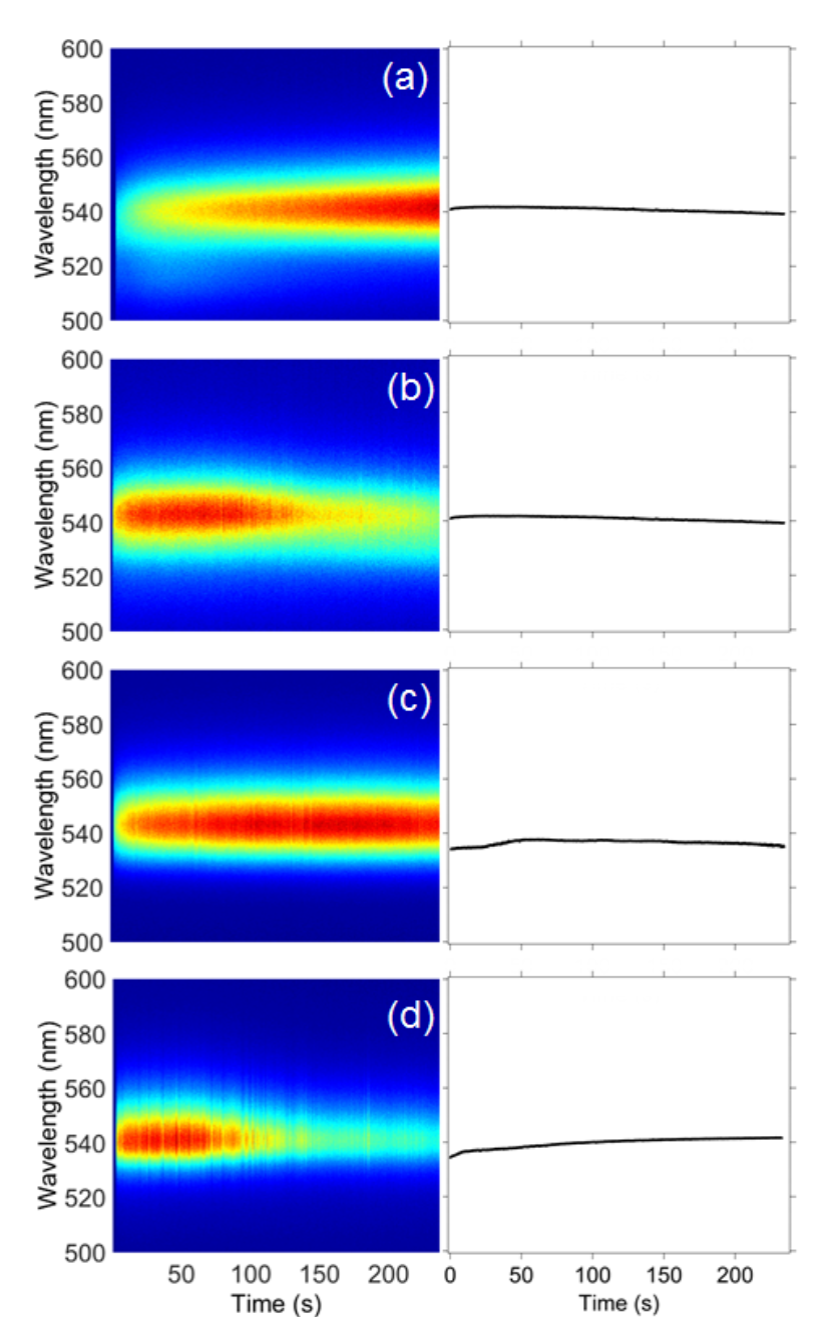
Fig. 8
Time-correlated luminescence microspectroscopy spectra of single CH3NH3PbBr3 perovskites. The left column shows the plots of luminescence versus illumination time with a 488 nm laser (1.58 × 105 W/cm2) for CH3NH3PbBr3 perovskites synthesized using Scheme 2. The samples are: (a) unwashed sample, (b) washed sample, (c) unwashed with excess precursor, and (d) washed with excess precursor sample. The right column shows the average λmax versus illumination time (n=3).
Any deviations in λmax are less than 4 nm, and can be explained as measurement uncertainty (e.g., minor changes to the focus). Hu et al. measured no shifts in the luminescence λmax for a single CsPbI3 nanocrystal after 600 s when excited using a pulsed laser at a temperature of 4 K [62]. They reported a resolution-limited peak width of about 200 µeV, but a laser power density was not reported that could be compared to the experimental parameters we used. Rainò et al. also observed a stable luminescence λmax for a mixed halide CsPb(Br/Cl)3 nanocrystal measured for 60 s at 6 K using 92 W/cm2 excitation [63]. Multiple luminescence peaks and a shifting λmax were measured at higher power densities (up to 4.7 × 103 W/cm2); the time-correlated luminescence behavior varied from one nanocrystal to another. In our study, nine of the CH3NH3PbI3 nanocrystals measured at room temperature exhibited stable time-correlated luminescence at a higher excitation power density than used by Rainò et al., while three showed a <79-nm shift in λmax. Eight of the CH3NH3PbBr3 nanocrystals we measured exhibited stable luminescence behavior while four showed a <8-nm shift in λmax.
To examine the stability of perovskites prepared using the optimized synthetic method with prolonged illumination, luminescence microspectroscopy of individual nanocrystals was employed. For this experiment, a batch of 24-hour-aged perovskites in toluene was divided into four samples. One sample was left as is, and is referred to as the “unwashed sample.” The second sample was mixed with additional 0.012-mM alkylammonium halide (i.e., the same concentration as the precursor solution) and was labeled “unwashed with excess precursor.” For the third and fourth samples, the perovskites were precipitated from the product solution and were resuspended in toluene (“washed sample”) or an 0.01- mM alkylammonium halide solution in toluene (“washed with excess precursor sample”).
The CH3NH3PbI3 perovskites prepared using Scheme 2 exhibited a constant λmax of 800 nm for all samples except the unwashed sample with excess precursor. For the latter, the λmax shifts from 737 nm at the start of illumination to 767 nm after 240 s of illumination. Thus, for CH3NH3PbI3 additional precursor affects the photostability whether it is present during the synthesis or added post synthesis, although the effect varies (e.g., blue-shifted peak versus red-shifted peak). While the λmax is stable for most of the CH3NH3PbI3 samples, the luminescence intensity is variable over the measurement period and photobrightening or photobleaching was measured for most of the nanocrystals (Fig. S2). We have previously reported and discussed this behavior [61].
The nanocrystalline CH3NH3PbBr3 perovskites prepared using the optimized synthesis (Scheme 2) exhibited a constant luminescence λmax around 540 nm (Fig. 8) for all samples, even when additional precursor was added to the washed nanocrystals. (No shifts of λmax greater than 4 nanometers were measured). Freppon et al. report the emission of these nanocrystals to be 498 nm when measured in toluene [61]. The red shift in λmax measured for the time-correlated luminescence spectra were measured in the dry state, which explains the difference in the λmax for these two studies. The fact that the luminescence λmax does not shift after adding excess precursor to the nanocrystals synthesized with the optimized method indicates that the higher precursor concentration must be present during the synthesis to have an effect on generating the photoinitiated luminescence peaks for CH3NH3PbBr3. Photobrightening and photobleaching were also recorded for CH3NH3PbBr3 nanocrystals (Fig. S3).
Conclusion
In order to be useful in a variety of applications, nanocrystalline perovskites need to be photostable. The non-optimized organometal halide perovskites show shifts in luminescence λmax and exhibit multiple luminescence peaks when excess alkylammonium precursor is used. The optimized synthetic method produces nanocrystalline methylammonium lead halide particles that are photostable over at least 4 minutes of focused illumination. The mechanism for the improved photostability is likely to be reduced surface traps when low precursor ratios are utilized. This work reports on the photostability of perovskites containing a single halide. Similar synthetic methods may increase the photostability of related perovskites. In the case of mixed-halide perovskite nanocrystals, where domains of heterogeneous halide compositions may exist, the photostability is more complicated. For example, our initial investigation has shown that CH3NH3Pb(Br0.2I0.8)3 nanocrystalline perovskites exhibit an abrupt shift in (λmax from 630 nm to 750 nm after a few seconds of illumination followed by a constant (λmax with additional illumination (Fig. S4). Under indentical conditions, our initial investigations indicate that single-halide perovskites exhibit stable (λmax as reported in this work. Thus, the continued study of mixed-halide perovskites should be pursued.
Acknowledgment
This research is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences through the Ames Laboratory. The Ames Laboratory is operated for the U.S. Department of Energy by Iowa State University under Contract No. DE-AC02-07CH11358. We thank Miles Arthur White for assistance with graphics.
References
1. Green, M. A.; Ho-Baillie, A. ACS Energy Letters. 2017, 2, 822-830
2. Li, Z.; Klein, T. R.; Kim, D. H.; Yang, M.; Berry, J. J.; van Hest, M. F.; Zhu, K. Nature Reviews Materials. 2018, 3, 18017
3. Sutherland, B. R.; Sargent, E. H. Nature Photonics. 2016, 10, 295
4. Dong, Q.; Fang, Y.; Shao, Y.; Mulligan, P.; Qiu, J.; Cao, L.; Huang, J. Science. 2015, 347, 967-970
5. Kojima, A.; Teshima, K.; Shirai, Y.; Miyasaka, T. Journal of the American Chemical Society. 2009, 131, 6050-6051
6. Liu, M.; Johnston, M. B.; Snaith, H. J. Nature. 2013, 501, 395
7. Burschka, J.; Pellet, N.; Moon, S.-J.; Humphry-Baker, R.; Gao, P.; Nazeeruddin, M. K.; Gratzel, M. Nature. 2013, 499, 316-319
8. Zhou, H.; Chen, Q.; Li, G.; Luo, S.; Song, T.-b.; Duan, H.-S.; Hong, Z.; You, J.; Liu, Y.; Yang, Y. Science. 2014, 345, 542-546
9. Yang, W. S.; Noh, J. H.; Jeon, N. J.; Kim, Y. C.; Ryu, S.; Seo, J.; Seok, S. I. Science. 2015, 348, 1234-1237
10. Yang, W. S.; Park, B.-W.; Jung, E. H.; Jeon, N. J.; Kim, Y. C.; Lee, D. U.; Shin, S. S.; Seo, J.; Kim, E. K.; Noh, J. H. Science. 2017, 356, 1376-1379
11. Christians, J. A.; Miranda Herrera, P. A.; Kamat, P. V. Journal of the American Chemical Society. 2015, 137, 1530-1538
12. Bert, C.; Jeroen, D.; Nicolas, G.; Aslihan, B.; Jan, D. H.; Lien, D. O.; Anitha, E.; Jo, V.; Jean, M.; Edoardo, M.; De, A. F.; Hans‐Gerd, B. Advanced Energy Materials. 2015, 5, 1500477
13. Berhe, T. A.; Su, W.-N.; Chen, C.-H.; Pan, C.-J.; Cheng, J.-H.; Chen, H.-M.; Tsai, M.-C.; Chen, L.-Y.; Dubale, A. A.; Hwang, B.-J. Energy & Environmental Science. 2016, 9, 323-356
14. Samrana, K.; Khaja, N. M.; Michael, G.; Shahzada, A. Angewandte Chemie International Edition. 2014, 53, 2812-2824
15. Gao, P.; Gratzel, M.; Nazeeruddin, M. K. Energy & Environmental Science . 2014, 7, 2448-2463
16. Dou, L.; Wong, A. B.; Yu, Y.; Lai, M.; Kornienko, N.; Eaton, S. W.; Fu, A.; Bischak, C. G.; Ma, J.; Ding, T. Science. 2015, 349, 1518-1521
17. Boix, P. P.; Agarwala, S.; Koh, T. M.; Mathews, N.; Mhaisalkar, S. G. The Journal of Physical Chemistry Letters. 2015, 6, 898-907
18. Sichert, J. A.; Tong, Y.; Mutz, N.; Vollmer, M.; Fischer, S.; Milowska, K. Z.; Garcia Cortadella, R.; Nickel, B.; Cardenas-Daw, C.; Stolarczyk, J. K. Nano Letters. 2015, 15, 6521-6527
19. Protesescu, L.; Yakunin, S.; Bodnarchuk, M. I.; Krieg, F.; Caputo, R.; Hendon, C. H.; Yang, R. X.; Walsh, A.; Kovalenko, M. V. Nano Letters. 2015, 15, 3692-3696
20. Deschler, F.; Price, M.; Pathak, S.; Klintberg, L. E.; Jarausch, D.-D.; Higler, R.; Huttner, S.; Leijtens, T.; Stranks, S. D.; Snaith, H. J.; Atature, M.; Phillips, R. T.; Friend, R. H. The Journal of Physical Chemistry Letters. 2014, 5, 1421-1426
21. Zhang, F.; Zhong, H.; Chen, C.; Wu, X.-g.; Hu, X.; Huang, H.; Han, J.; Zou, B.; Dong, Y. ACS Nano. 2015, 9, 4533-4542
22. Zhu, F.; Men, L.; Guo, Y.; Zhu, Q.; Bhattacharjee, U.; Goodwin, P. M.; Petrich, J. W.; Smith, E. A.; Vela, J. ACS Nano. 2015, 9, 2948-2959
23. Yang, Z.; Zhang, W.-H. Chinese Journal of Catalysis. 2014, 35, 983-988
24. Stranks, S. D.; Nayak, P. K.; Zhang, W.; Stergiopoulos, T.; Snaith, H. J. Angewandte Chemie International Edition. 2015, 54, 3240-3248
25. Yan, K.; Long, M.; Zhang, T.; Wei, Z.; Chen, H.; Yang, S.; Xu, J. Journal of the American Chemical Society. 2015, 137 (13), 4460-4468
26. Tan, Z.-K.; Moghaddam, R. S.; Lai, M. L.; Docampo, P.; Higler, R.; Deschler, F.; Price, M.; Sadhanala, A.; Pazos, L. M.; Credgington, D.; Hanusch, F.; Bein, T.; Snaith, H. J.; Friend, R. H. Nature Nanotechnology. 2014, 9, 687-692
27. Zhang, M.; Yu, H.; Lyu, M.; Wang, Q.; Yun, J.-H.; Wang, L. Chemical Communications. 2014, 50, 11727-11730
28. Jang, D. M.; Park, K.; Kim, D. H.; Park, J.; Shojaei, F.; Kang, H. S.; Ahn, J.-P.; Lee, J. W.; Song, J. K. Nano Letters. 2015, 15, 5191-5199
29. Noh, J. H.; Im, S. H.; Heo, J. H.; Mandal, T. N.; Seok, S. I. Nano Letters. 2013, 13, 1764-1769
30. Dimesso, L.; Dimamay, M.; Hamburger, M.; Jaegermann, W. Chemistry of Materials. 2014, 26, 6762-6770
31. Misra, R. K.; Aharon, S.; Li, B.; Mogilyansky, D.; Visoly-Fisher, I.; Etgar, L.; Katz, E. A. The Journal of Physical Chemistry Letters. 2014, 326-330
32. Hoke, E. T.; Slotcavage, D. J.; Dohner, E. R.; Bowring, A. R.; Karunadasa, H. I.; McGehee, M. D. Chemical Science. 2015, 6, 613-617
33. Sadhanala, A.; Deschler, F.; Thomas, T. H.; Dutton, S. E.; Goedel, K. C.; Hanusch, F. C.; Lai, M. L.; Steiner, U.; Bein, T.; Docampo, P.; Cahen, D.; Friend, R. H. The Journal of Physical Chemistry Letters. 2014, 5, 2501-2505
34. Gottesman, R.; Gouda, L.; Kalanoor, B. S.; Haltzi, E.; Tirosh, S.; Rosh-Hodesh, E.; Tischler, Y.; Zaban, A.; Quarti, C.; Mosconi, E. The Journal of Physical Chemistry Letters. 2015, 6, 2332-2338
35. Shao, Y.; Xiao, Z.; Bi, C.; Yuan, Y.; Huang, J. Nature Communications. 2014, 5, 5784
36. Yuan, Y.; Huang, J. Accounts of Chemical Research. 2016, 49, 286-293
37. Stranks, S. D.; Burlakov, V. M.; Leijtens, T.; Ball, J. M.; Goriely, A.; Snaith, H. J. Physical Review Applied. 2014, 2, 034007
38. Wu, X.; Trinh, M. T.; Niesner, D.; Zhu, H.; Norman, Z.; Owen, J. S.; Yaffe, O.; Kudisch, B. J.; Zhu, X.-Y. Journal of the American Chemical Society. 2015, 137, 2089-2096
39. Wehrenfennig, C.; Liu, M.; Snaith, H. J.; Johnston, M. B.; Herz, L. M. The Journal of Physical Chemistry Letters. 2014, 5, 1300-1306
40. Lee, M. M.; Teuscher, J.; Miyasaka, T.; Murakami, T. N.; Snaith, H. J. Science. 2012, 338, 643-647
41. Agiorgousis, M. L.; Sun, Y.-Y.; Zeng, H.; Zhang, S. Journal of the American Chemical Society. 2014, 136, 14570-14575
42. Sangni, W.; Furong, H.; Liya, Z.; Jianwu, W.; Youling, X.; Peng, J.; Zhuo, C.; Zuodong, Y.; Qi, P.; Zhong, Z. J. ChemNanoMat. 2018, 4, 409-416
43. Yin, W.-J.; Shi, T.; Yan, Y. Applied Physics Letters. 2014, 104, 063903
44. Wan‐Jian, Y.; Tingting, S.; Yanfa, Y. Advanced Materials. 2014, 26, 4653-4658
45. Kim, J.; Lee, S.-H.; Lee, J. H.; Hong, K.-H. The Journal of Physical Chemistry Letters. 2014, 5, 1312-1317
46. Shi, T.; Yin, W.-J.; Hong, F.; Zhu, K.; Yan, Y. Applied Physics Letters. 2015, 106, 103902
47. Buin, A.; Pietsch, P.; Xu, J.; Voznyy, O.; Ip, A. H.; Comin, R.; Sargent, E. H. Nano Letters. 2014, 14, 6281-6286
48. Abate, A.; Saliba, M.; Hollman, D. J.; Stranks, S. D.; Wojciechowski, K.; Avolio, R.; Grancini, G.; Petrozza, A.; Snaith, H. J. Nano Letters. 2014, 14, 3247-3254
49. Noel, N. K.; Abate, A.; Stranks, S. D.; Parrott, E. S.; Burlakov, V. M.; Goriely, A.; Snaith, H. J. ACS Nano. 2014, 8, 9815-9821
50. Xu, J.; Buin, A.; Ip, A. H.; Li, W.; Voznyy, O.; Comin, R.; Yuan, M.; Jeon, S.; Ning, Z.; McDowell, J. J. Nature Communications. 2015, 6, 7081
51. Bhattacharjee, U.; Freppon, D.; Men, L.; Vela, J.; Smith, E. A.; Petrich, J. W. ChemPhysChem. 2017, 18 (18), 2526-2532
52. Grabolle, M.; Spieles, M.; Lesnyak, V.; Gaponik, N.; Eychmuller, A.; Resch-Genger, U. Analytical Chemistry. 2009, 81, 6285-6294
53. Schmidt, L. C.; Pertegas, A.; Gonzalez-Carrero, S.; Malinkiewicz, O.; Agouram, S.; Minguez Espallargas, G.; Bolink, H. J.; Galian, R. E.; Perez-Prieto, J. Journal of the American Chemical Society. 2014, 136, 850-853
54. De Roo, J.; Ibanez, M.; Geiregat, P.; Nedelcu, G.; Walravens, W.; Maes, J.; Martins, J. C.; Van Driessche, I.; Kovalenko, M. V.; Hens, Z. ACS Nano. 2016, 10, 2071-2081
55. Yoon, S. J.; Stamplecoskie, K. G.; Kamat, P. V. The Journal of Physical Chemistry Letters. 2016, 7, 1368-1373
56. Rosales, B. A.; Hanrahan, M. P.; Boote, B. W.; Rossini, A. J.; Smith, E. A.; Vela, J. ACS Energy Letters. 2017, 2, 906-914
57. Fedeli, P.; Gazza, F.; Calestani, D.; Ferro, P.; Besagni, T.; Zappettini, A.; Calestani, G.; Marchi, E.; Ceroni, P.; Mosca, R. The Journal of Physical Chemistry C. 2015, 119, 21304-21313
58. Tanaka, K.; Takahashi, T.; Ban, T.; Kondo, T.; Uchida, K.; Miura, N. Solid State Communications. 2003, 127, 619-623
59. Pellet, N.; Teuscher, J.; Maier, J.; Gratzel, M. Chemistry of Materials. 2015, 27, 2181-2188
60. Stranks, S. D.; Eperon, G. E.; Grancini, G.; Menelaou, C.; Alcocer, M. J. P.; Leijtens, T.; Herz, L. M.; Petrozza, A.; Snaith, H. J. Science. 2013, 342, 341-344
61. Freppon, D. J.; Men, L.; Burkhow, S. J.; Petrich, J. W.; Vela, J.; Smith, E. A. Journal of Materials Chemistry C. 2017, 5, 118-126
62. Hu, F., Yin, C., Zhang, H., Sun, C., Yu, W.W., Zhang, C., Wang, X., Zhang, Y. and Xiao, M., Nano Letters. 2016. 16(10), 6425-6430
63. Raino, G., Nedelcu, G., Protesescu, L., Bodnarchuk, M.I., Kovalenko, M.V., Mahrt, R.F., Stoferle, T. ACS Nano. 2016 10(2), 2485-2490.
Author notes
*Corresponding author: Emily A. Smith, e-mail: esmith1@iastate.edu; Javier Vela, e-mail: vela@iastate.edu
