Journal of the Mexican Chemical Society, vol. 63, no. 2, 2019
Sociedad Química de México A.C.
Santiago García
Universidad de Guanajuato, Mexico
Karla Armendariz
Universidad de Guanajuato, Mexico
Tayde Villaseñor-Granados
Universidad de Guanajuato, Mexico
Patricia Ponce-Noyola
Universidad de Guanajuato, Mexico
Alberto Flores
Universidad de Guanajuato, Mexico
José Ascención Martínez-Álvarez
Universidad de Guanajuato, Mexico
Marco A. García-Revilla
Universidad de Guanajuato, Mexico
Juvencio Robles
Universidad de Guanajuato, Mexico
Yolanda Alcaraz
Universidad de Guanajuato, Mexico
Miguel A. Vázquez *
Universidad de Guanajuato, Mexico
Date received: 29 November 2018
Date accepted: 23 April 2019
Funding
Funding source: DAIP-UG
Contract number: 811/2016
Funding
Funding source: CONACYT
Contract number: 328700
Contract number: 229121
Abstract.: Due to increasing drug resistance by Candida species, especially in hospitals, the search for new antifungal agents has intensified. The incorporation of the coumarin scaffold into several nitrogen-containing heterocyclic moieties reportedly increases antimicrobial efficiency. The aim of this study was to design and synthesize a series of simple coumarin-linked triazole derivatives and test their possible antifungal activity against four Candida species. Docking simulations were conducted to explore the binding properties of the test compounds and compare them to reported data on fluconazole, the reference drug. Starting from 3-acetylcoumarins, coumarins 2a-d, 3a-c and 4a-d were obtained in high yields. The concentration of each compound needed to inhibit the Candida species was determined by serial dilution. An inhibition of 62% of C. albicans was produced by 2b (300 µg/ml), 87% of C. tropicalis by 3a (100 µg/ml), 89% of C. parapsilosis by 3a (500 µg/ml), and 87% of C. glabrata by 4a (300 µg/ml). The values of antifungal activity were similar for the coumarin derivatives and fluconazole, the latter of which induced 90% inhibition of the four yeasts at 500 µg/ml. According to the docking simulations, the interactions at the active site of the lanosterol 1,4-demethylase enzyme (CYP51) are similar for the test compounds and fluconazole. The subcellular location of the derivatives was identified as the mitochondrion. These coumarins are characterized by structural simplicity, with the simplest structures showing better antifungal activity than fluconazole. Further research is needed to isolate CYP51 and directly test its inhibition by coumarin derivatives.
Keywords: Simple coumarin-azide, Coumarin-triazole, CYP51, Antifungal activity, Candida yeasts, Docking.
Resumen.: Una serie de moléculas de cumarina-triazol se sintetizaron y evaluaron contra diferentes especies de Candida. Las cumarinas 2a-d, 3a-c y 4a-d se obtuvieron utilizando como material de partida las 3-acetilcumarinas en altos rendimientos. La concentración necesaria de las moléculas para mostrar actividad antifúngica contra las cuatro especies de Candida se determinó mediante un método de diluciones seriadas. Se reporta un 62% de inhibición de C. albicans usando 2b (300 µg/ml), 87% de inhibición contra C. parapsilosis por 3a (500 µg/ml), y un 87% de inhibición a C. glabrata por 4a (300 µg/ml). El efecto de las cumarinas es comparado con el fármaco de referencia fluconazol, que induce un 90% de inhibición en todas las cepas usando 500 µg/ml. Los resultados del estudio Docking muestran que las interacciones de todas las moléculas en el sitio activo de la enzima CYP51 son similares a las interacciones presentadas por el fluconazol. Finalmente, tomando ventaja de las propiedades fluorescentes de las cumarinas, la localización subcelular y penetración de los compuestos, fue localizada en las mitocondrias. Las cumarinas reportadas, además de presentar sencillez estructural, también presentan valores de inhibición de las cepas comparables, y en los casos mencionados, mejores que el fármaco de referencia.
Palabras clave: Cumarina-azida, Derivados cumarina-triazol, CYP51, Actividad Antifúngica, Levaduras de Candida, Docking, fluorescencia.
Introduction
The coumarin moiety is present in the molecular structure of bioactive drugs used against a wide range of diseases. [1] It forms part of compounds showing antibacterial, [2] antifungal, [3] anti-inflammatory, [4] analgesic, [5] anticancer, [6] anti-HIV, [7] antiviral [8] and anticoagulant activity. [9,10] Consequently, the coumarin structure is considered a privileged scaffold in medicinal chemistry [11] and is included in many antifungal compounds (Fig. 1A). Many research groups have modified the molecular structure of coumarin in an attempt to improve its biological activity. [7,10,12]
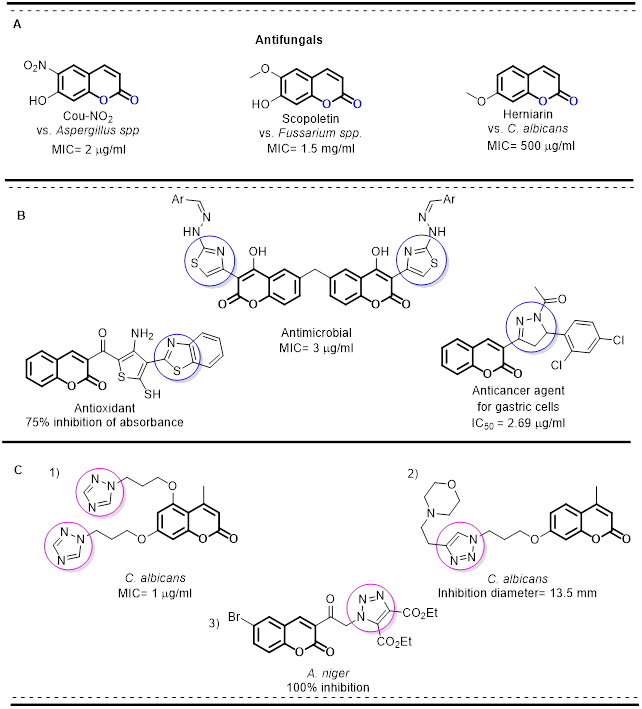
Fig. 1
A) The structures of some drugs containing a coumarin scaffold; B) substitution patterns on the coumarin moiety and the resulting activity; C) coumarin-triazole molecules as antifungal compounds.
Patients who have undergone a stem cell transplant, antineoplasic chemotherapy, an organ transplant or an HIV infection are targets for invasive opportunistic fungal infections. This susceptibility together with increasing fungal resistance to treatment has led to high morbidity and mortality rates.[13-15] Indeed, a growing number of fungal species are becoming multidrug resistant, in the majority of cases due to long-term drug therapy.[16] Since a large number of antifungal drugs on the market are now of limited effectiveness (and some imply serious risks), there is an urgent need for the design, synthesis and potentiation of novel antifungal agents.
The azole moiety also has a broad spectrum of activity against yeasts and filamentous fungi. [17-19] Its accepted mechanism of action is the inhibition of lanosterol 14 α-demethylase (the CYP51 enzyme), [20,21] a part of the cytochrome P450 family. This enzyme catalyzes the demethylation of lanosterol, a precursor of ergosterol (the main component of the fungal cellular membrane). Hence, any damage to it causes fungal death.[22]
According to reports in recent years, the incorporation of the coumarin scaffold into several nitrogen-containing heterocyclic moieties (Fig. 1B) increases antimicrobial efficiency and broadens the spectrum of bioactivity (Fig. 1C). [23,24] The substituents on the coumarin ring play an important role in modifying biological activity. Depending on the substituent, pharmacological characteristics related to toxicity and therapeutic activity can be dramatically different. [25,26] The inclusion of the coumarin moiety in the synthesis of pharmacophores has allowed for the development of novel molecules.
The aim of the present study was to synthesize a series of simple coumarin-based molecules with a 1,2,3-triazole, a 1,2,4-triazole or -Br as a substituent on the coumarin moiety, and then test their possible antifungal activity against four Candida yeast species. The final subcellular location of the molecules was identified with fluorescent imaging. Docking simulations were carried out on Mycobacterium tuberculosis (MT)CYP51 to analyze the binding properties of the coumarin derivatives. The findings were compared to the reported binding properties of the reference drug, fluconazole.
Experimental
Chemistry
All solvents and reagents were of commercial grade and used without further purification. Melting points were determined on an RY-1 MP apparatus. Absorption spectra were recorded on a Perkin-Elmer Lambda 50 apparatus, and emission spectra were recorded on an FL-7000 spectrometer. ESI-HRMS spectra were recorded on a Maxis Impact ESI-QTOF-MS spectrometer and Bruker Daltonics mass spectrometer. 1H and 13C NMR spectra were recorded in CDCl3 or DMSO-d6 solutions on the Bruker Avance III HD, either with the Bruker Ascent 400 MHz magnet or with the Bruker Ultra Shield 500 MHz magnet. NMR procedures were performed at 25 °C with TMS and assigned solvent signals as internal standards. Chemical shifts are expressed in ppm (δ) and the J constant in Hertz.
General procedure for preparing 3-acetyl-coumarin derivatives
The salicylaldehyde derivative (1 mmol), morpholine (0.1 mmol) and ethyl acetoacetate (1.2 mmol) were dissolved in 10 mL of ethanol and the mixture was subjected to microwave irradiation at 140 °C (140 W) for 5 min.[27] Water was added and the precipitate filtered to furnish the desired coumarin (85-90%).
3-acetylcoumarin. Yellow solid; mp 107-109 °C. IR(KBr) νmax/cm-1= 1741, 1678; UV-vis (CH2Cl2) λmax/nm= 301; NMR 1H (500 MHz, CDCl3) δ 8.52 (s, 1H), 7.66 (d, J = 2.8 Hz, 2H), 7.41-7.33 (m, 2H), 2.74 (s, 3H); NMR 13C (125 MHz, CDCl3) δ= 195.5, 159.2,155.3, 147.4, 134.3, 130.2,125.0, 124.9, 118.3, 116.7, 30.5.
6-methoxy-3-acetylcoumarin. Yellow needles; mp 177-179 °C. IR(KBr) νmax/cm-1= 1731, 1680; UV-vis (CH2Cl2) λmax/nm= 327; NMR 1H (500 MHz, CDCl3) δ 8.46 (s, 1H), 7.29 (t, J = 8.1 Hz, 1H), 7.23 (d, J = 9.0 Hz, 1H), 7.04 (s, 1H), 3.87 (s, 4H), 2.73 (s, 3H); NMR 13C (125 MHz, CDCl3) δ= 195.57, 159.37, 156.39, 149.96, 147.19, 124.72, 122.83, 118.55, 117.75, 111.18, 55.90, 30.51.
6-bromo-3-acetylcoumarin. White solid; mp 218-222 °C. IR (KBr) νmax/cm-1= 1735, 1675; UV-vis (CH2Cl2) λmax/nm= 270; NMR 1H (500 MHz, CDCl3) δ 8.41 (s, 1H), 7.78 (s, 1H), 7.73 (d, J = 8.6 Hz, 1H), 7.26 (s, 1H), 2.73 (s, 3H); NMR 13C (125 MHz, CDCl3) δ= 194.9, 158.52, 154.11, 145.90, 137.01, 132.18, 125.50, 119.75, 118.42, 117.52, 30.47.
7-dietilamino-3-acetylcoumarin. Yellow solid; mp 150-151 °C. IR (KBr) νmax/cm-1= 1725, 1664; UV-vis (CH2Cl2) λmax/nm= 433; Em (CH2Cl2) λmax/nm= 467; NMR 1H (400 MHz, CDCl3) δ 8.43 (s, 1H), 7.39 (d, J = 9.0 Hz, 1H), 6.62 (dd, J = 9.0, 2.3 Hz, 1H), 6.47 (d, J = 1.9 Hz, 1H), 3.46 (q, J = 7.1 Hz, 4H), 2.68 (s, 3H), 1.25 (t, J = 7.1 Hz, 6H); NMR 13C (100 MHz, CDCl3) δ= 195.74, 160.88, 158.77, 153.02, 147.86, 131.89, 116.18, 109.85, 108.18, 96.60, 45.15, 30.59, 12.44.
General procedure for preparing 3-(2-bromoacetyl)-coumarin derivatives 1a-d [27]
A 3-acetylcoumarin derivative (1 mmol) was dissolved in CH3CN (2 ml), followed by the addition of trifluoroacetic acid (2 mmol) and N-bromosuccinimide (NBS, 1.1 mmol). The mixture was subjected to microwave irradiation at 140 °C for 15 min, then extracted three times by using CH2Cl2. The combined organic extract was dried and subsequently recrystallized with EtOH to afford 1a-d. The NMR analysis of these coumarins matched published reports on the same compounds.[28]
1a. Brown solid, 95% yield; mp 163-164 °C. IR (KBr) (max/cm-1= 1727, 1685, 1613, 1602; UV-vis (CH2Cl2) λmax/nm= 247; NMR 1H (400 MHz, CDCl3) δ 8.64 (s, 1H), 7.71 (t, J = 7.2 Hz, 2H), 7.44-7.35 (m, 2H), 4.76 (s, 2H); NMR 13C (100 MHz, CDCl3) δ 188.90, 158.88, 155.43, 149.54, 135.12, 130.43, 125.31, 122.19, 118.15, 116.91, 35.59.
1b. Yellow solid, 88% yield; mp 213-215 °C. IR (KBr) (max/cm-1= 1730, 1689, 1605; UV-vis (CH2Cl2) λmax/nm= 246; NMR 1H (500 MHz, CDCl3) δ 8.59 (s, 1H), 7.33 (d, J = 8.6 Hz, 1H), 7.28 (d, J = 9.9 Hz, 1H), 7.07 (s, 1H), 4.75 (s, 2H), 3.88 (s, 3H); NMR 13C (125 MHz, CDCl3) δ 188.98, 156.62, 150.10, 149.24, 123.71, 122.37, 118.45, 118.02, 111.11, 55.98, 35.62.
1c. Brown solid, 91% yield; mp 184-185 ºC. IR (KBr) ν max/cm-1= 1730, 1687; UV-vis (CH2Cl2) λmax/nm= 294; NMR 1H (500 MHz, CDCl3) δ 8.53 (s, 1H), 7.83 (d, J = 2.3 Hz, 1H), 7.78 (dd, J = 8.8, 2.3 Hz, 1H), 7.30 (d, J = 8.8 Hz, 1H), 4.72 (s, 2H); NMR 13C (125 MHz, CDCl3) δ 188.6, 158.2, 154.2, 148.0, 137.7, 132.3, 123.2, 119.6, 118.6, 117.9, 35.2.
1d. Red solid, 60% yield; mp 211-213 °C. IR (KBr) (max/cm-1= 1729, 1686; UV-vis (CH2Cl2) λmax/nm= 460; Em (CH2Cl2) λmax/nm=474; NMR 1H (400 MHz, CDCl3) δ 8.54 (s, 1H), 7.51 - 7.40 (m, 1H), 6.66 (dd, J = 9.0, 2.0 Hz, 1H), 6.47 (s, 1H), 4.75 (s, 2H), 3.48 (c, J = 7.1 Hz, 4H), 1.26 (t, J = 7.1 Hz, 6H); NMR 13C (100 MHz, CDCl3) δ= 190.94, 160.72, 158.95, 153.65, 148.95, 132.33, 113.09, 110.30, 108.38, 96.62, 45.31, 36.93, 12.45.
General procedure for preparing 3-(2-azidoacetyl)-coumarin derivatives 2a-d
These derivatives were prepared with a modified version of the procedure described by Kusanur. [29] Potassium azide (1.2 mmol) was dissolved in 5 ml of water before slowly adding the solution to a previously formed mixture of 1a-d (1 mmol) dissolved in dioxane (4 ml). After stirring the reaction at room temperature (rt) for 1 h, 2 ml of water were added, and the precipitate was filtered and recrystallized with ethanol to give the corresponding pure compound 2a-d.
3-(2-azidoacetyl)-coumarin (2a). Brown solid, 80% yield; mp 136-137 ºC. IR (KBr) νmax/cm-1= 2113, 1727,1690; UV-vis (CH2Cl2) λmax/nm= 307; NMR 1H (400 MHz, CDCl3) δ 8.70 (s, 1H), 7.72 (t, J = 6.3 Hz, 2H), 7.40 (t, J = 7.8 Hz, 2H), 4.74 (s, 2H); NMR 13C (125 MHz, CDCl3) δ 191.19, 159.05, 155.43, 149.26, 135.28, 130.61, 125.37, 122.03, 118.09, 116.90, 58.70.
6-methoxy-3-(2-azidoacetyl)-coumarin (2b). Yellow solid, 70% yield; mp 138-139 ºC. IR (KBr) νmax/cm-1= 2112, 1712, 1566; UV-vis (CH2Cl2) λmax/nm= 315.90; NMR 1H (500 MHz, CDCl3) δ 8.65 (s, 1H), 7.34 (d, J = 8.9 Hz, 1H), 7.30 (s, 1H), 7.08 (s, 1H), 4.74 (s, 2H), 3.88 (s, 3H); NMR 13C (125 MHz, CDCl3) δ 191.30, 159.23, 156.68, 150.99, 150.09, 149.07, 123.99, 117.99, 111.26, 58.74, 55.97.
6-bromo-3-(2-azidoacetyl)-coumarin (2c). Brown solid, 70% yield; mp 147-149 °C. IR (KBr) νmax/cm-1= 2115, 1686, 1608, 1554; NMR 1H (500 MHz, CDCl3) δ 8.61 (s, 1H), 7.87 (d, J = 1.3 Hz, 1H), 7.81 (dd, J = 8.8, 1.4 Hz, 1H), 7.32 (d, J = 8.8 Hz, 1H), 4.74 (s, 2H); NMR 13C (125 MHz, CDCl3) δ 190.90, 154.16, 147.79, 137.92, 132.54, 122.93, 119.49, 118.60, 118.00, 58.67.
7-(diethylamino)-3-(2-azidoacetyl)-coumarin (2d). Orange solid, 60% yield; mp 162-163 ºC. IR (KBr) νmax/cm-1= 2016, 1711, 1613, 1569; UV-vis (CH2Cl2) λmax/nm= 445; Em (CH2Cl2) λmax/nm=474; NMR 1H (500 MHz, CDCl3) δ 8.55 (s, 1H), 7.44 (d, J =9.2, 1H), 6.66 (dd, J = 9.0, 2.0 Hz, 1H), 6.47 (s, 1H), 4.68 (s, 2H), 3.48 (q, J = 7.1 Hz, 5H), 1.26 (t, J = 7.1 Hz, 8H,); NMR 13C (125 MHz, CDCl3) δ 190.94, 160.72, 158.95, 153.65, 148.95, 132.33, 113.09, 110.30, 108.38, 96.62, 58.62, 45.31, 12.45.
General procedure for the preparation of 3-(2-(4-trimethylsilyl)-1H-1,2,3-triazol-1-yl) acetyl)-coumarin derivatives 3a-c
Derivatives 2a-d (1 mmol) were dissolved in 2 ml of CH2Cl2 in a round bottom flask before adding ethynyltrimethylsilane (1.2 mmol), sodium ascorbate (0.4 mmol) and CuCl2 (0.05 mmol). Then, 2 ml of water were placed in the flask and stirring began. [30] The reaction proceeded at rt for 6 h to provide 3a-c. Finally, the pure products were obtained by means of recrystallization from ethanol.
3-(2-(4-trimethylsilyl)-1H-1,2,3-triazol-1-yl)acetyl)-coumarin (3a). Brown solid, 70% yield; mp 157-159 °C. IR (KBr) νmax/cm-1= 2958, 1740, 1699, 1560; UV-vis (CH2Cl2) λmax/nm= 309; NMR 1H (500 MHz, CDCl3) δ 8.65 (s, 1H), 7.72 (s, 2H), 7.66 (s, 1H), 7.42 (d, J = 7.3 Hz, 2H), 6.00 (s, 2H), 0.35 (s, 9H); NMR 13C (125 MHz, CDCl3) δ 189.99, 160.38, 156.59, 150.89, 136.69, 131.89, 126.62, 119.14, 118.07, 59.38, 0.00.
6-methoxy-3-(2-(4-trimethylsilyl)-1H-1,2,3-triazol-1-yl) acetyl)-coumarin (3b). Yellow solid, 85% yield; mp 194-195 ºC. IR (KBr) νmax/cm-1= 2956, 1726, 1609, 1566; UV-vis (CH2Cl2) λmax/nm= 383.10; NMR 1H (500 MHz, CDCl3) 8.63 (s, 1H), 7.68 (s, 1H), 7.35 (d, J = 12.5 Hz, 2H), 7.09 (s, 1H), 6.01 (s, 2H), 3.88 (s, 3H), 0.40 (s, 9H); NMR 13C (125 MHz, CDCl3) δ 188.67, 159.46, 156.79, 150.21, 149.71, 124.49, 118.32, 118.10, 111.32, 58.57, 56.00, -1.06.
7-(diethylamino)-3-(2-(4-trimethylsilyl)-1H-1,2,3-triazol-1-yl)acetyl)-coumarin (3c). Yellow crystals, 60% yield; mp 172-173 °C. IR (KBr) νmax/cm-1= 2958, 1717, 1687, 1616, 1577; UV-vis (CH2Cl2) λmax/nm= 446; Em (CH2Cl2) λmax/nm=476; NMR 1H (500 MHz, CDCl3) δ 8.50 (s, 1H), 7.62 (s, 1H), 7.43 (d, J = 9.0 Hz, 1H), 6.66 (dd, J = 9.0, 2.4 Hz, 1H), 6.50 (d, J = 2.3 Hz, 1H), 5.96 (s, 2H), 3.50 (q, J = 7.1 Hz, 4H), 1.27 (t, J = 7.1 Hz, 6H), 0.35 (s, 9H); NMR 13C (125 MHz, CDCl3) δ 190.24, 160.19, 155.00, 150.27, 132.12, 111.54,109.51, 97.83, 59.37, 46.43, 13.53, -0.00.
General procedure for preparing 3-(2-(1H-1,2,4-triazol-1-yl) acetyl)-coumarin derivatives 4a-d
A mixture of one of the compounds 1a-d (1 mmol) and 1H-1,2,4-triazole (1.2 mmol) was dissolved in 2 ml dioxane. The reaction was initiated with the help of a stirring bar and took place at rt for 6 h. The resulting precipitate was filtered and washed with water to generate the corresponding pure product 4a-d.
3-(2-(1H-1,2,4-triazol-1-yl) acetyl)-coumarin (4a). White solid, 80% yield; mp 233-234 ºC; IR (KBr) νmax/cm-1= 2920, 1729, 1607, 1559; NMR 1H (500 MHz, DMSO) δ 9.44 (s, 1H), 8.97 (s, 1H), 8.10 (dd, J = 7.8, 1.5 Hz, 1H), 7.86 (ddd, J = 8.7, 7.4, 1.6 Hz, 1H), 7.57 (d, J = 8.4 Hz, 1H), 7.50 (td, J = 7.6, 1.0 Hz, 1H), 5.98 (s, 2H); NMR 13C (125 MHz, DMSO) δ 188.81, 159.16, 155.29, 150.01, 145.08, 136.20, 131.94, 125.91, 121.75, 118.47, 116.87, 56.16.
6-methoxy-3-(2-(1H-1,2,4-triazol-1-yl) acetyl)-coumarin (4b). Yellow solid, 80% yield; mp 234-235 ºC. IR (KBr) νmax/cm-1= 2843, 1721, 1620, 1567; NMR 1H (500 MHz, DMSO) δ 9.22 (d, J = 3.5 Hz, 2H), 8.89 (s, 1H), 7.65 (d, J = 2.9 Hz, 1H), 7.56-7.48 (m, 1H), 7.48-7.41 (m, 1H), 5.90 (s, 2H), 3.84 (s, 3H); NMR 13C (125 MHz, DMSO) δ 188.98, 159.29, 156.54, 149.83, 145.04, 145.04, 124.21, 121.90, 118.92, 118.02, 113.19, 56.42, 56.03.
6-bromo-3-(2-(1H-1,2,4-triazol-1-yl) acetyl)-coumarin (4c). White solid, 80% yield; mp 172-174 ºC. IR (KBr) νmax/cm-1= 2928, 1725, 1604, 1554; NMR 1H (500 MHz, DMSO) δ 9.12 (s, 2H), 8.88 (s, 1H), 8.35 (d, J = 2.4 Hz, 1H), 7.99 (dd, J = 8.9, 2.4 Hz, 1H), 7.54 (d, J = 8.9 Hz, 1H), 5.87 (s, 2H); NMR 13C (125 MHz, DMSO) δ 189.20 158.60, 148.39, 138.12, 133.58, 122.93, 120.32, 119.14, 117.41, 55.49.
7-(diethylamino)-3-(2-(1H-1,2,4-triazol-1-yl) acetyl)-coumarin (4d). Yellow solid, 60% yield; mp 198-200 ºC. IR (KBr) νmax/cm-1= 2974, 1708, 1612, 1505; UV-vis (CH2Cl2) λmax/nm= 442; Em (CH2Cl2) λmax/nm=476; NMR 1H (500 MHz, DMSO) δ 9.25-9.14 (m, 2H), 8.66 (s, 1H), 7.76 (d, J =9.0 Hz, 1H), 6.88 (d, J = 8.9 Hz, 1H), 6.66 (s, 1H), 5.80 (s, 2H), 3.53 (q, J = 6.9 Hz, 4H), 1.17 (t, J = 6.9 Hz, 6H).
Biological assays
All compounds were dissolved in DMSO and tested at various concentrations against each of the four Candida species: C. albicans, C. tropicalis, C. parapsilosis and C. glabrata. The antifungal activity of the coumarin derivatives was determined with the serial dilution method. Fluconazole (500 (g/mL) served as the standard drug.
The activity against each (human) pathogenic yeast was evaluated in solid medium. Briefly, 1 x 106 cells of each Candida species were grown in 1 mL of YPD broth together with one of the coumarin derivatives (at the corresponding concentration) at 37 °C for 16 h in an orbital shaker at 120 rpm. Subsequently, serial dilutions were made (1:2000 final dilution) to inoculate three microliters of each dilution over YPD plates and carry out incubation at 28 °C for 16 h. Upon completion of this period, the colony-forming units (CFUs) were counted to calculate the percentage of inhibition (with DMSO as the control) by using a variation of the method described by Sanglard.[29]
Fluorescence assay
To conduct fluorescence microscopy, the Candida cells cultured in YPD medium were collected by centrifugation at 10,000 rpm for 2 min. The supernatant was removed, the cells were resuspended in 100 μL PBS, and 10 μL of a 10-5 M solution of one of the compounds (1c, 2d, 3c or 4d) was added. Following incubation at 25 °C for 5 min, another centrifugation was done at 10,000 rpm for 2 min. The cells were resuspended in PBS and placed on a glass slide, to be examined with fluorescence microscopy at a wavelength of 380 nm and an emission of 420 nm.
To obtain the mitochondrial fraction, 3 mL of the yeast culture was centrifuged at 10,000 rpm for 2 min. Then one volume of glass beads was added and agitated to break the cells. The cell extract was centrifuged at 2,500 rpm for 5 min and the cell-free extract at 12,000 rpm for 10 min. The pellet was resuspended in PBS and centrifuged at 5,500 rpm for 5 min to eliminate the nuclei. The supernatant was centrifuged at 12,000 rpm for 10 min. After resuspending the mitochondria in 100 μL of PBS, 50 µL of the sample were mixed with 2 µL of probe dissolved in DMSO. Upon completing 5 min of incubation, the sample was centrifuged at 10,000 rpm, the pellet resuspended in PBS, and fluorescence observed on a confocal microscope (at 63X).
Computational studies
The Molegro Virtual Docker (MVD) version 2011.5.0 [31] was employed to visualize, manipulate and dock the MTCYP51 protein structure, and to perform the molecular docking calculations. The protein was kept rigid and the positions of all its residues were fixed to reduce the computational effort. The conformation of the ligands was allowed to vary during the simulation, leaving the dihedral angles free for the minimization of the total binding energy.
Results and discussion
Chemistry
The main interactions of fluconazole with the active site of a CYP51 enzyme (herein Mycobacterium tuberculosis) have been described. [32] The cavity of the active site was divided into 4 main subregions (indicated by red, gray, green and blue), characterized by the types of functional groups able to form interactions with fluconazole (Fig. 2). [33] The analysis of the structure of fluconazole (left) and of a generalized version of the present coumarins reveals similarities (Fig. 3). The aromatic ring of fluconazole can be substituted by a coumarin moiety (marked in red), and the two triazole functional groups of fluconazole (marked in blue, attached to the acetyl group) could have properties similar to just one triazole ring, an azide or a bromine of the coumarins.
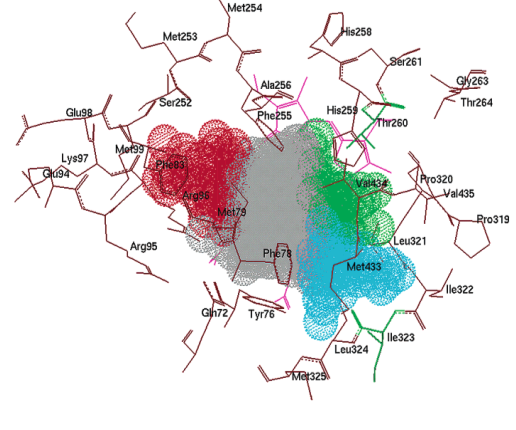
Fig. 2
Illustration of the different zones of the CYP51 active site.
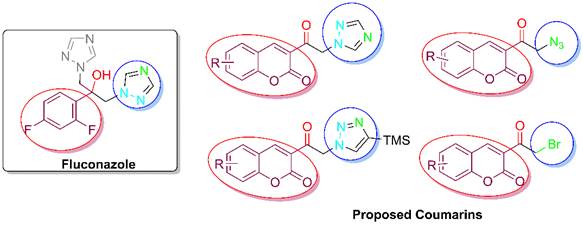
Fig. 3
Structural comparison between fluconazole and the coumarin scaffold.
One of the triazole rings of fluconazole interacts with the active site subregions containing an Fe atom (Fig. 2, blue and green). The same type of interaction can be seen with the different functional group moieties of the coumarins. For instance, the second triazole ring interacts with the gray subregion. Even though not all the proposed compounds generate such interactions, a challenge in the present study was to explore the relevance of this nucleus to biological activity. The red section of the structures may be located in the hydrophobic funnel of the active site entrance, in relation to which both fluconazole and the coumarins exhibit the same behavior. Finally, the fluconazole hydroxyl group displays hydrophilic interactions with the Arg96 and Gln72m residues, also observed with the oxygen of the coumarin carbonyl moiety.
Bearing in mind that the proposed coumarins show structural similarity, the simpler ones may have the desired antifungal activity. Hence, an attempt was herein made to establish a suitable route for their synthesis. A strategy was proposed for synthesizing a simple functionalized compound (Scheme 1) chosen to test the biological activity of the molecules that could fit into the active site. The molecules were divided into four sets (Fig. 4), depending on the presence of a common functional group or pharmacophore selected for its antifungal activity. Since the structures of the molecules of each set are similar, a comparable binding mode on the biological target was expected. Each of the four molecules in set 1 contains an α-bromated carbonyl group. The Br atom was replaced by an azide in set 2, while the azide group was cyclized to obtain a 1,2,3-triazole ring in set 3. In set 4, each of the molecules has a 1,2,4-triazole ring.
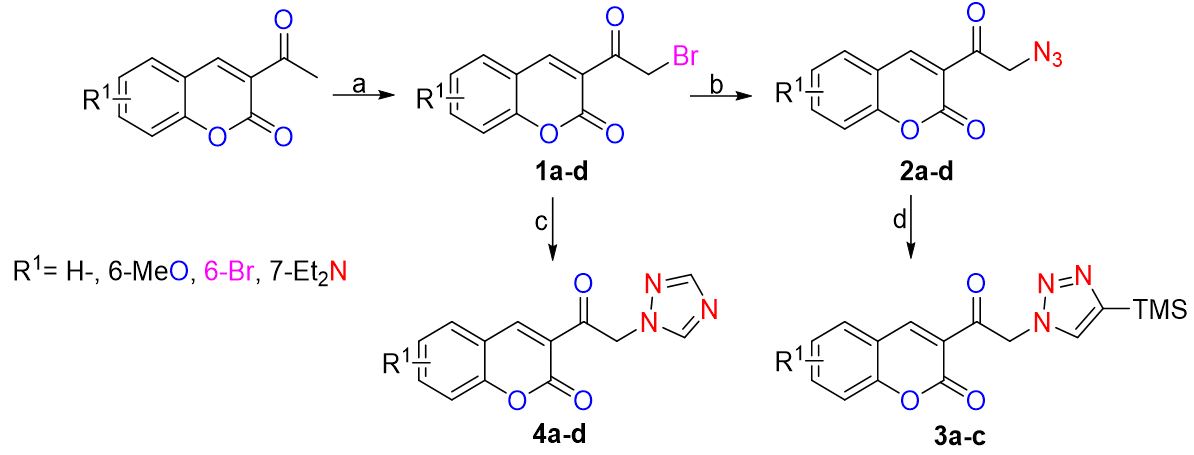
Scheme 1
Synthetic route for the test compounds.
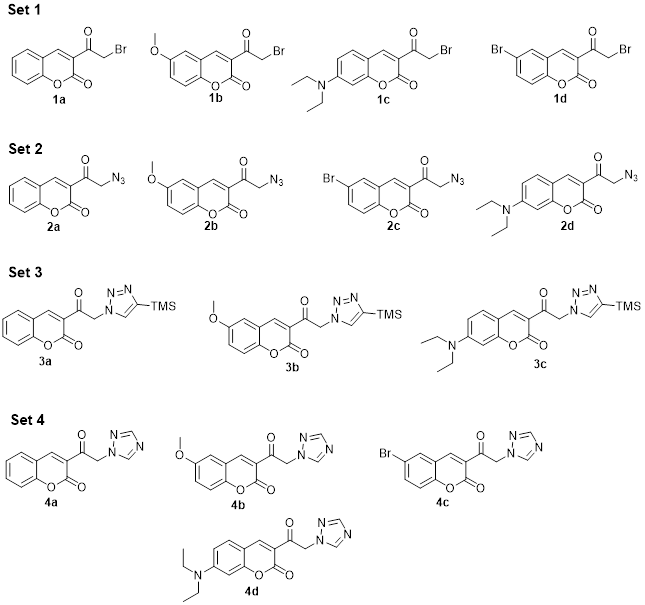
Fig. 4
Molecular structures of the four series of synthesized compounds.
The starting coumarins were prepared by a previously reported procedure. [34] The 3-acetylcoumarins were synthesized by Knoevenagel’s condensation between the 1,3-dicarbonilic compound and the corresponding salicylaldehyde, with morpholine as a base. The characteristic signals displayed in the 1H NMR of the compounds are a displacement from 8.41 to 8.46 ppm and from 2.68 to 2.73 ppm, attributed to the vinylic proton in position 4 and the methyl, respectively. On the other hand, the 13C NMR spectra contain signals at 147.0 and 30 ppm, respectively. The IR spectra includes a band of 1730 cm-1, characteristic of the ketone in position 3 of coumarins.
The methyl signal serves as an indicator of the conversion to α-halogenated compounds. There were different methodologies implemented to achieve this conversion, such as the addition of Br2 in acid medium and CuBr2 under neutral conditions. Although the desired compounds were afforded, it was difficult to control the reaction due to the presence of mono-, di- and tri- halogenation products. The coumarins were isolated with a chromatographic column and a 1:1 CH2Cl2/hexane system, leading to low yields.
However, the same compounds could be obtained in good yields and without chromatography by adopting the methodology in Lee's report, [28] with NBS as a halogenating agent and under acidic conditions. 1H NMR spectra demonstrate a shift of the α-halogenated methylene signal to 4.72-4.76 ppm for the expected products, as well as the integration of 2H. With 13C NMR, a displacement of 35 ppm is shown for this signal. IR spectra do not provide relevant information for these products.
The 1H NMR spectra for 1a-d and 2a-d had signals from 4.68 ppm to 4.74 ppm, characteristic of a methylene substituted with an azide group. The 13C NMR spectra reveal the corresponding methylene signal in a high field displacement in regard to the halogenated precursor, from 35 ppm to 58 ppm. The IR spectra exhibited a band at 2100 cm-1, attributed to the azide group. Next, click chemistry was applied by using ethynyltrimethylsilane to generate only one regioisomer (3a-c), causing the methylene signal in 1H NMR to be displaced to higher fields, (4.7 ppm to 6 ppm) due the electronic effect of the triazole ring. Consequently, the IR band of the azide disappeared, indicating the complete transformation of the starting materials.
As an alternative pathway, a halogenated ketone (1 a-d) was dissolved in dioxane and heated to 60 °C. Subsequently, 1H-1,2,4-triazole was added to furnish the desired compound (4a-d) as a precipitate (Scheme 1). 1H NMR spectra of the set 4 molecules have signals from 5.80 ppm to 5.98 ppm, characteristic of a methylene substituted with the 1,2,4-triazole. 13C NMR spectra evidence a displacement of methylene to 55-56 ppm.
A UV-Vis study of the molecules was conducted to determine the maximum absorption and thus choose the best molecule for fluorescence microscopy. The effect of various solvents on the absorption of 10-6 M 3-acetylcoumarin was analyzed to understand the structure-behavior relationship when using the same concentration of the compound. With acetone, the spectrum displays a notable band overlap. There were solubility problems with hexanes and wide bands with toluene. Despite the minimal overlap observed with acetonitrile, not all the compounds are soluble in this solvent. The best solvent for obtaining the maximum absorption turned out to be CH2Cl2 (see SI). An overlap of the normalized absorption spectra of 3-acetylcoumarin is illustrated for the different solvents. As can be appreciated, the maximum absorption band is slightly displaced, and the intensity of absorption is dependent on the solvent (Fig. 5).
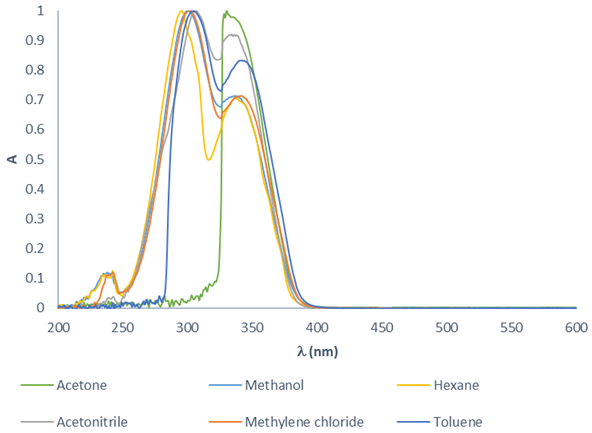
Fig. 5
Overlapping of the normalized UV spectra of 3-acetyl coumarin, recorded with various solvents.
To complete this part of the study, an examination was made of the effect on maximum absorption caused by the group in position 3 and the substituent in the aromatic moiety. The overlap of the normalized absorption spectra of compounds 1-4a (Fig. 6) reveal no significant displacement of the maximum absorption, not even by the bromide or the triazole moiety. Except for compound 4a, which has the lowest solubility, the spectra show the same behavior for all test compounds. Since the series of coumarin derivatives with the diethylamine substituent (1c, 2d, 3c and 4d) exhibited a bathochromic shift, none of these compounds could be employed for the fluorescence analysis.
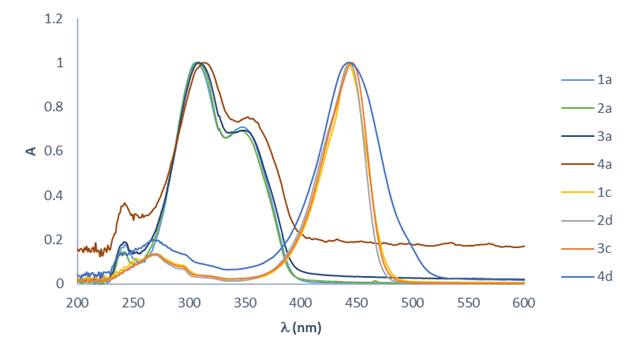
Fig. 6
Comparison of the UV spectra of two families of coumarins (R = H-, and 7-Et2N-) in CH2Cl2 (10-6 M).
Once the wavelength of maximum absorption was known, the maximum emission length was evaluated. Afterward, it could be established whether each compound had a large enough Stokes shift to be used in fluorescence microscopy. Accordingly, a comparison was made between the absorption spectra and the emission spectra of the family of diethylamino coumarins (Fig. 7), finding that the shifts were not significant, the average value of all the compounds being 50 nm. However, this value was large enough to assure that the compounds would not undergo auto-absorption.
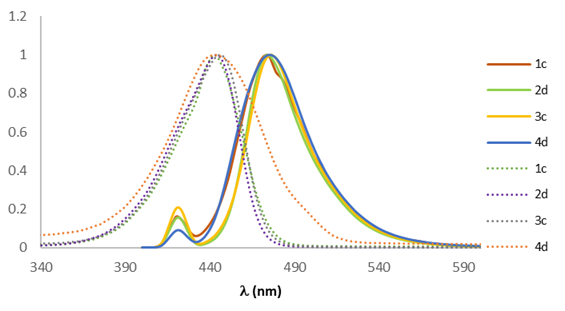
Fig. 7
Comparison of the absorption (dotted line) and the emission (solid line) spectra of fluorescent coumarins, determined in CH2Cl2 (10-6 M).
Evaluation of antifungal activity
All four Candida species exhibited sensitivity to one or more compounds (Table 1). C. albicans was sensitive to 1a, 2a, 3a, 4a and 4c, C glabrata to 1a, 2a, 3a and 2b, C. parapsilosis to 3a, and C. tropicalis to 1a, 2a, 4a, 1b and 2b. Compounds 2a and 3a were the most effective against all the Candida species analyzed. C. parapsilosis proved to be the most resistant to the antifungal activity of the test compounds. The present results provide insights into promising alternatives for the treatment of candidiasis.
Concentration of the coumarin compounds and their inhibition of Candida.
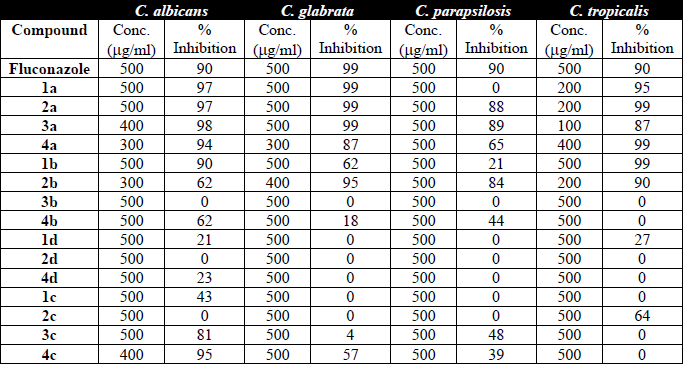
The inhibitory concentration of fluconazole is 500 μg/ml for diverse fungi, similar to the MIC of the present coumarin derivatives. The differences in activity of the compounds against the four Candida species could stem from the interaction between the coumarin derivatives and the CYP51 enzyme. Further research is needed on this interaction by using the isolated CYP51 enzyme. Whereas 2a and 3a showed the best experimental activity on all four yeast species, corresponding to a ligand efficiency (LE) value very similar to that of fluconazole, 2d and 3b did not produce an antifungal effect against any of the species examined (even at high concentrations).
Fluorescence imaging
The evaluation of all compounds by UV-visible spectroscopy revealed a maximum absorption of about 240 nm with CHCl3 and MeOH (1x10-5 M). Fluorescence microscopy of C. albicans displayed the accumulation of the test compounds in the living yeast cells. Compound 2d was chosen for this study due to its better solubility, fluorescent properties and lower antifungal activity. Fluorescence imaging illustrates the presence of 2d inside cell mitochondria (Fig. 8), which may be attributed to the coumarin core (Table 1).
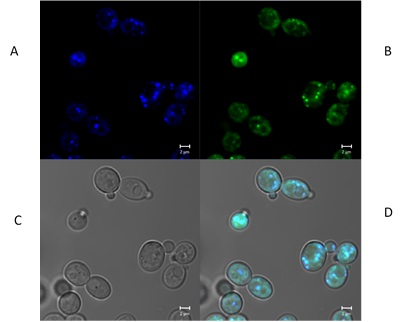
Fig. 8
C. albicans cells were viewed under a microscope (at 63X): A) with UV light and a DAPI filter (380 nm), B) with UV light and a FITSI filter (580 nm), and C) in bright field. D) The images were merged. The fluorescent mitochondria of 2d appear in the images made with both filters.
The fact that 2d was observed in mitochondria could explain its lack of biological activity, because most enzymes in the ergosterol biosynthesis pathway are concentrated in the endoplasmic reticulum.[35] On the other hand, 2a was one of the most effective compounds, demonstrating the importance of the substituent in the coumarin core. Coumarin and azide seem to play a key role in the antifungal activity of the compounds.
The compounds that have been found in the mitochondria of C. albicans and C. glabrata reportedly cause mitochondrial dysfunction as well as elevated respiratory activity and efflux levels, thus contributing to the susceptibility of these yeasts to azoles.[36] Moreover, the cell mitochondrion is likely to affect an antifungal drug response, in part due to its role in membrane lipid homeostasis.[37]
To clearly establish that the light spots in the image (Fig. 8) represent mitochondria, the organelles were purified from a cell culture. The coumarins were able to effectively target the organelles by using probes, leading to direct damage to cell function and metabolism (Fig. 9).

Fig. 9
Stained mitochondria isolated from compound 2d under a fluorescence microscope equipped with a DAPI filter.
Docking
Applying a previously reported methodology,[33] docking simulations were carried out to compare the binding modes of the test and reference compounds. By analyzing the five most probable cavities in the MTCYP51 target, the largest was found to be the active site. For each of the coumarin derivatives herein evaluated, a blind docking calculation was performed to identify the preferred binding site within the MTCYP51 protein and compare it to that of fluconazole. All these derivatives bind to the active site of the enzyme. Subsequently, a specific docking calculation was made inside the active site. The binding modes shown (Fig. 10) correspond to the best pose obtained for each molecule in set 1 and for fluconazole.
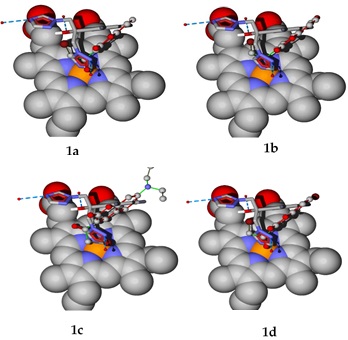
Fig. 10
Binding modes inside the MTCYP51 active site are illustrated for each molecule in set 1 of the coumarin derivatives (ball and stick model) and for fluconazole (stick model). The binding modes of sets 2-4 are similar to those of set 1 (see SI).
The binding energy and LE were calculated for the best pose of each molecule in set 1 (Table 1, SI). Compound 1a, with a binding affinity of -7.11 kcal/mol.atom towards the MTCYP51 active site, most closely approached the LE of -7.3 kcal/mol.atom previously described for fluconazole.33 Compounds 1b and 1d also exhibit good affinity towards the MTCYP51 active site, as can be inferred from their LE values. A subtle but very important difference was observed for the binding mode of 1c compared to the other three set 1 molecules.
The compounds most similar to fluconazole in terms of their LE value (Table 1) and binding mode (Fig. 9, SI) are 4a and 4b (LE = -7.11 and -7.34 kcal/mol.atom, respectively, vs -7.31 kcal/mol.atom). The coumarin moiety in both 4a and 4b is perfectly aligned with the bifluorinated phenyl ring of fluconazole at the hydrophobic S3 subregion of MTCYP51. In addition, the triazole rings in 4a, 4b and fluconazole are perfectly aligned and oriented towards the Fe atom of the heme group, creating a strong electrostatic interaction (~ -15.30 kcal/mol). This suggests the likely coordination with the Fe atom.
Furthermore, molecules 4a, 4b and fluconazole form similar hydrogen bonds. The triazole ring of each of these molecules interacts with the 2087 water molecule located at the S1 hydrophilic subregion of MTCYP51. Moreover, the carbonyl oxygen on the coumarin moiety in both 4a and 4b forms a hydrogen bond, like the one established by fluconazole between its hydroxyl group and the 2175 water molecule located at the S4 hydrophilic subregion of MTCYP51. The two isosteric groups (the carbonyl oxygen atom on the coumarin moiety in both 4a and 4b and the hydroxyl group of fluconazole) are also perfectly aligned.
Another insight provided by the docking results is the low LE value of 1c and 4c, which corresponds to their limited antifungal activity determined experimentally. It is necessary to carry out assays with the isolated CYP51 enzyme to examine whether coumarin derivatives inhibit its function. The LE and the dose needed to trigger antifungal activity were similar for 4a, 3d and the reference drug. Depending on the drug and the Candida species, the mechanism of antifungal resistance may be inherent or can be acquired after a period of exposure. All Candida species resistant to multiple antifungal drugs [38] are susceptible to fluconazole, although a dose-dependent effect has only been detected for C. glabrata. Some of the coumarin derivatives are just as effective as fluconazole for inhibiting the growth of the four Candida species included in the study.
Conclusion
A series of coumarin derivatives was synthesized with two kinds of triazole moieties (1,2,3- and 1,2,4-triazoles) and tested on four Candida species. Most of the new compounds showed significantly lower antifungal activity than fluconazole towards three of the yeast cultures. Whereas the strongest resistance to most compounds was produced by C. parapsilosis, a considerable effect against this yeast was generated by 2a and 3a, which were also efficient inhibitors of C. glabrata. C. Tropicalis was sensitive towards 1a, 2a, 4a, 1b and 2b. In general, the greatest antifungal activity was against C. albicans, with the best results afforded by 1a, 2a, 3a and 4a. In these compounds, the coumarin group does not have substituents on the aromatic ring. The present findings offer new insights into the design and development of simple structures of antifungal drugs. Such structures do not require a second triazole ring since with only one ring they had biological activity similar to that of the reference drug.
In vivo fluorescence imaging demonstrated that the compounds entered the mitochondrion after passing through the lipidic membrane. Consequently, the mechanism of action for antifungal activity could consist of inhibiting lanosterol-α-demethylase and/or directly damaging the mitochondrion. Further research is needed to clarify this question.
According to the docking simulations, there is no significant difference between the test compounds in terms of their manner of interaction at the active site of the biological target. Hence, they share the functional groups responsible for an adequate binding mode. For some of the coumarins (e.g., 2b, 3b and 4a), molecular coupling with the MTCYP51 enzyme took place with an LE value similar to that of fluconazole. However, it is necessary to experimentally evaluate the interaction between the coumarin derivatives and the isolated CPY51 enzyme. The simpler coumarin structures herein tested caused an antifungal effect comparable to that of the reference drug, fluconazole.
Acknowledgements
The authors are grateful for the financial support provided by the Dirección de Apoyo a la Investigación and the Posgrado (DAIP-UG; grant 811/2016). We thank Bruce A. Larsen for proofreading. SG and TVG appreciate the support from CONACYT scholarships (328700 and 229121). The authors are thankful to the Laboratorio Nacional de Caracterización de Propiedades Fisicoquímicas y Estructura Molecular (University of Guanajuato-CONACYT; project 260373) for the computing time and analytical data provided. We also acknowledge Karla Gisela Armendariz (Project 241803) for technical support in regard to the fluorescence test.
References
1. Wu, L.; Wang, X.; Xu, W.; Xu, F. F. and R. Current Medicinal Chemistry. 2009, pp 4236-4260.
2. Arshad, A.; Osman, H.; Bagley, M. C.; Lam, C. K.; Mohamad, S.; Zahariluddin, A. S. M. Eur. J. Med. Chem. 2011.
3. Renuka, N.; Ajay Kumar, K. Bioorganic Med. Chem. Lett. 2013, 23 (23), 6406-6409.
4. Bansal, Y.; Sethi, P.; Bansal, G. Med. Chem. Res. 2013, 22 (7), 3049-3060.
5. Keri, R. S.; Sasidhar, B. S.; Nagaraja, B. M.; Santos, M. A. Eur. J. Med. Chem. 2015, 100, 257-269.
6. Dalla Via, L.; Gia, O.; Marciani Magno, S.; Santana, L.; Teijeira, M.; Uriarte, E. J. Med. Chem. 1999, 42 (21), 4405-4413.
7. Kumar, R.; Saha, A.; Saha, D. Fitoterapia 2012, 83 (1), 230-233.
8. Hwu, J. R.; Singha, R.; Hong, S. C.; Chang, Y. H.; Das, A. R.; Vliegen, I.; De Clercq, E.; Neyts, J. Antiviral Res. 2008, 77 (2), 157-162.
9. Gomez-Outes, A.; Suarez-Gea, M. L.; Calvo-Rojas, G.; Lecumberri, R.; Rocha, E.; Pozo-Hernandez, C.; Vargas-Castrillon, A. I. T.-F. and E. Current Drug Discovery Technologies. 2012, pp 83-104.
10. Zhang, R.-R.; Liu, J.; Zhang, Y.; Hou, M.-Q.; Zhang, M.-Z.; Zhou, F.; Zhang, W.-H. Eur. J. Med. Chem. 2016, 116, 76-83.
11. Welsch, M. E.; Snyder, S. A.; Stockwell, B. R. Curr. Opin. Chem. Biol. 2010, 14 (3), 347-361.
12. Carpinella, M. C.; Ferrayoli, C.; Palacios, S. M. J. Agric. Food Chem. 2005, 53, 2922-2927.
13. Ji, Q.; Ge, Z.; Ge, Z.; Chen, K.; Wu, H.; Liu, X.; Huang, Y.; Yuan, L.; Yang, X.; Liao, F. Eur. J. Med. Chem. 2016, 108, 166-176.
14. Pfaller, M. A.; Diekema, D. J. Clin. Microbiol. Rev. 2007, 20 (1), 133-163.
15. Thati, B.; Noble, A.; Rowan, R.; Creaven, B. S.; Walsh, M.; McCann, M.; Egan, D.; Kavanagh, K. Toxicol. Vitr. 2007, 21 (5), 801-808.
16. Bowyer, P.; Moore, C. B.; Rautemaa, R.; Denning, D. W.; Richardson, M. D. Curr. Infect. Dis. Rep. 2011, 13 (6), 485.
17. Jeu, L.; Piacenti, F. J.; Lyakhovetskiy, A. G.; Fung, H. B. Clin. Ther. 2003, 25 (5), 1321-1381.
18. Pfaller, M. A.; Messer, S. A.; Hollis, R. J.; Jones, R. N. Antimicrob. Agents Chemother. 2001, 45 (10), 2862-2864.
19. Shaikh, M. H.; Subhedar, D. D.; Khan, F. A. K.; Sangshetti, J. N.; Shingate, B. B. Chinese Chem. Lett. 2015, 27 (2), 295-301.
20. Lepesheva, G. I.; Zaitseva, N. G.; Nes, W. D.; Zhou, W.; Arase, M.; Liu, J.; Hill, G. C.; Waterman, J. Biol. Chem. 2006, 281 (6), 3577-3585.
21. Verras, A.; Alian, A.; Ortiz De Montellano, P. R. Protein Eng. Des. Sel. 2006, 19 (11), 491-496.
22. W., R. V.; F., S. T.; Luciana, T.; V., S. M.; C., C. H.; R., R. C.; A., A. P. Fundam. Clin. Pharmacol. 2016, 31 (1), 37-53.
23. Gouda, M. A.; Berghot, M. A.; Baz, E. A.; Hamama, W. S. Med. Chem. Res. 2012, 21 (7), 1062-1070.
24. Raghu, M.; Nagaraj, A.; Reddy, C. S. J. Heterocycl. Chem. 2009, 46 (2), 261-267.
25. Musa, M. A.; Cooperwood, J. S.; Khan, M. O. F. Pharmacotherapy of Breast Cancer.; 2008; Vol. 15.
26. Muhammad Asif. Chem. Int. 2015, 1 (1), 1-11.
27. García, S.; Vázquez, J. L.; Rentería, M.; Aguilar-Garduño, I. G.; Delgado, F.; Trejo-Durán, M.; García-Revilla, M. A.; Alvarado-Méndez, E.; Vázquez, M. A. Opt. Mater. (Amst). 2016, 62, 231-239.
28. Lee, J. C.; Bae, Y. H.; Chang, S. K. Bull. Korean Chem. Soc. 2003, 24 (4), 407-408.
29. Kusanur, R. A.; Kulkarni, M. V. Indian J. Chem. - Sect. B Org. Med. Chem. 2005, 44 (3), 591-594.
30. Kushwaha, K.; Kaushik, N.; Lata; Jain, S. C. Design and Synthesis of Novel 2H-Chromen-2-One Derivatives Bearing 1,2,3-Triazole Moiety as Lead Antimicrobials; 2014; Vol. 24.
31. Thomsen, R.; Christensen, M. H. J. Med. Chem. 2006, 49 (11), 3315-3321.
32. Ji, H.; Zhang, W.; Zhang, M.; Kudo, M.; Aoyama, Y.; Yoshida, Y.; Sheng, C.; Song, Y.; Yang, S.; Zhou, Y.; et al. J. Med. Chem. 2003, 46 (4), 474-485.
33. Villaseñor-Granados, T.; García, S.; Vazquez, M. A.; Robles, J. Theor. Chem. Acc. 2016, 135 (9), 210.
34. Rentería Gómez, M.; López Vallejo, F. I.; Alcaraz Contreras, Y.; Flores Martínez, A.; Martínez Rosales, J. M.; Vázquez Guevara, M. Acta Univ. 2011, 21 (Regular), 74-81.
35. Mense, S. M.; Zhang, L. Cell Res. 2006, 16, 681.
36. Shingu-Vazquez, M.; Traven, A. Eukaryot. Cell 2011, 10 (11), 1376-1383.
37. Benhamou, R. I.; Bibi, M.; Steinbuch, K. B.; Engel, H.; Levin, M.; Roichman, Y.; Berman, J.; Fridman, M. ACS Chem. Biol. 2017, 12 (7), 1769-1777.
38. Silva, S.; Negri, M.; Henriques, M.; Oliveira, R.; Williams, D. W.; Azeredo, J. FEMS Microbiol Rev 2012, 36, 288-305.
Author notes
*Corresponding author: Miguel A. Vázquez, e-mail: mvazquez@ugto.mx Department of Chemistry, DCNyE, University of Guanajuato, Guanajuato, México; Tel/Fax: +52 (473) 732 0006, Ext. 1419.
