Journal of the Mexican Chemical Society, vol. 63, no. 1, 2019
Sociedad Química de México A.C.
Juan Carlos Martínez-Hernández
Universidad Autónoma Metropolitana, Mexico
Iris N. Serratos
Universidad Autónoma Metropolitana, Mexico
César Millán-Pacheco
Facultad de Farmacia, Universidad Autónoma del Estado de Morelos, México., Mexico
Arturo Rojo-Domínguez
Universidad Autónoma Metropolitana, Mexico
Jaqueline Padilla-Zúñiga *
Universidad Autónoma Metropolitana, Mexico
Date received: 06 September 2018
Date accepted: 12 February 2019
Funding
Funding source: National Council of Science and Technology (CONACYT, Mexico)
Contract number: 194059
Abstract: Human cathepsins K, L, and S, which are involved in the development of several serious diseases, are strongly inhibited by their related prosegments, to which they are covalently bound or simply forming complexes. In this work, three-dimensional structures of the three natural complexes of these enzymes with their related proregions were constructed, as well as six chimeric complexes of the same three prosegments with their non-cognate enzymes. We made a comparative study of the contacts in all nine structures throughout their active sites. The analysis was performed looking for a structural parameter that could agree with the values of the inhibition constants reported experimentally for each of the nine complexes. We found that this correlating parameter was the difference of the electrostatic energy (involving hydrogen bonds and ion pairs) at the binding interface of a 13-amino acid fragment of the prosegments. We used the results of this work, on the one hand, to identify the key residues involved in the electrostatic intermolecular recognition in each studied complex and, on the other, to explain some results achieved by different research groups on the inhibition of the same enzymes analyzed here. It was found that the natural cathepsin L complex showed a higher number of electrostatic interactions, some of them interconnected, when compared to the other two natural complexes. In addition, the chimeric contacts revealed binding sites that could be used to achieve a more potent inhibition of these cathepsins, avoiding cross-interactions.
Keywords: human cathepsins, cathepsin inhibition, proteinase chimeric complexes, protease prosegments, cysteine proteases.
Resumen: Se sabe que las catepsinas humanas K, L y S, involucradas en el desarrollo de diversas enfermedades graves, son fuertemente inhibidas por sus prosegmentos cognados, a los que se unen covalentemente o sólo formando complejos. En este trabajo se construyeron las estructuras tridimensionales de los tres complejos naturales de estas enzimas, con sus prorregiones cognadas, así como seis complejos quiméricos de los mismos tres prosegmentos con enzimas no cognadas. Todo lo anterior se cumplió para llevar a cabo un estudio comparativo de los contactos estructurales de la interfaz a lo largo del sitio activo enzimático en los complejos construidos. El análisis implicó la búsqueda de un parámetro estructural que correlacionara con las constantes de inhibición reportadas experimentalmente para los nueve complejos estudiados. Se encontró que este parámetro fue la diferencia de energía electrostática (debida a enlaces de hidrógeno y pares iónicos) en la unión al sitio activo de un fragmento de 13 aminoácidos de los prosegmentos. Usamos los resultados de este trabajo, por una parte, para identificar los residuos clave involucrados en el reconocimiento intermolecular en cada complejo estudiado y, por otra, para explicar algunos datos reportados por diferentes grupos de investigación sobre la inhibición de las mismas enzimas aquí analizadas. Se encontró que el complejo natural de catepsina L establece un mayor número de interacciones electrostáticas, algunas de ellas altamente interconectadas, en comparación con los otros dos complejos naturales. Además, los contactos quiméricos revelaron sitios de unión que podrían usarse para lograr una inhibición más potente de estas catepsinas, evitando interacciones cruzadas.
Palabras clave: catepsinas humanas, inhibición de catepsinas, complejos proteínicos quiméricos, prosegmentos de proteasas, proteasas cisteínicas.
Abbreviations
CK, mature cathepsin K; CL, mature cathepsin L; CS, mature cathepsin S; pCK, prosegment of cathepsin K; pCL, prosegment of cathepsin L; pCS, prosegment of cathepsin S; MD, molecular dynamics; PDB, protein data bank; Ki, inhibition constant; vdw, van der Waals.
Introduction
Human cathepsins K, L and S have been targets for the development of a large number of inhibitors because they are involved in the progression of degenerative or lethal diseases, such as osteoporosis and cancer metastasis [1-10]. These proteases are synthesized as inactive precursors, or zymogens, with an N-terminal segment of approximately 100 amino acids covalently bonded to the enzyme chain. This segment folds as an independent structural domain and is called a prosegment or proregion, and it attaches to the active site with inhibition constants in the nanomolar range [11-13]. To acquire their biological activity, these enzymes must remove the proregion following a series of processing steps that yield the completely free and functional mature enzyme [14-17]. Crystallographic structures of the precursors of these three cathepsins have been determined and are available from the Protein Data Bank [18].
Prosegments bind the cleft of the proteases active site in cavities and areas denoted by S and S´ symbols, according to the notation of Berger and Schechter [19] and reviewed later by Turk et al. [20]. The experimental inhibition constants (Ki) of the natural and chimeric enzyme-prosegment complexes for cathepsins K, L and S were reported by Guay et al. [21] and then collected by Wiederanders in his review [22]. In the present work we denote a natural complex when the cathepsin is inhibited by its own prosegment, and chimeric when a prosegment of a cathepsin inhibits a different enzyme. In both natural and chimeric cases, the proregion of each cathepsin inhibits the other enzymes by forming non-covalent complexes, that is, without a peptide bond between the C-terminus of the prosegment and the N-terminus of the enzyme. Although the experimental inhibition data has been available for some time, a comparative molecular description to analyze the interactions of these proteases with their cognate and chimeric prosegments has not yet been carried out. All this information could be useful for the development of better and more specific inhibitors as in the case of the related enzyme cathepsin B [23,24].
In this paper, we present the construction of three-dimensional models of the natural complexes of cathepsins K, L and S with their cognate prosegments, pCKCK, pCLCL and pCSCS, respectively, and the six chimeric complexes, pCKCL, pCKCS, pCLCK, pCLCS, pCSCK and pCSCL. These human cathepsins were selected because besides the reported Ki values of the natural and chimeric prosegment-enzyme complexes, above mentioned, both the structures of their precursors and their complexes with organic inhibitors have been published in the PDB. All this data allowed us to construct the prosegment-enzyme complexes by homology modeling, and then to find a measurable structural parameter related to experimental values. This quantitative parameter of interaction was searched by its better correlation with the experimentally reported inhibition constants. Intermolecular van der Waals and electrostatic contact energies were calculated for each complex interface. The latter parameter, for a 13-amino acid prosegment fragment, was selected because it better describes the Ki values tendency, while maintaining a short chain representative of organic enzyme inhibitors. Then this structural parameter was used to analyze the enzyme-prosegment recognition interface, residue by residue, along the active site cleft for each constructed complex.
In order to validate the way of binding of the modeled prosegments, we analyzed and compared the most energetic contacts observed for the proregions with those established in the three-dimensional structures, deposited in the PDB, of specific inhibitors bonded to the corresponding three human cathepsins. Finally, our findings were used to offer structural explanation to some results obtained for other search groups.
Experimental
Materials and Methods
Molecular modeling and optimization of 3D prosegment-cathepsin structures
All visualization, molecular modeling, energy calculations and minimizations were accomplished with the Molecular Operating Environment (MOE) package [25] using AMBER99, a force field parameterized for protein molecules, unless otherwise stated. All missing hydrogen atoms were assigned with MOE; partial electric charges for each atom were estimated by the Gasteiger method [26]. The three prosegment-cathepsin complexes formed by their natural, non-chimeric sequences (pCKCK, pCLCL and pCSCS) were modeled from the crystallographic structures of their single chain precursors that were deposited in the Protein Data Bank with IDs: 1BY8 [27], 1CS8 [28] and 2C0Y [29], respectively. For this task, missing residues in the PDB files were added to match with sequences reported in UniProt [30] for the cathepsin K, L and S precursors, with access codes: CATK_HUMAN [31], CATL1_HUMAN [32] and CATS_HUMAN [33], respectively. Next, the peptide bond linking the prosegment with its mature enzyme moiety was eliminated, and the new N- and C-terminal groups were added, followed by a local geometry regularization until an RMS value of energy gradient below 40 kJ/(mol·nm) was reached. Modeling the 3D structures of the six chimeric complexes (pCKCL, pCKCS, pCLCK, pCLCS, pCSCK and pCSCL), for which no crystallographic information is available, made use of the HHpred server [34] based on the corresponding prosegment and mature cathepsin sequences, using the precursor structures of cathepsins K, L or S, as necessary. The proper stereochemical quality of all resulting structural models was validated by Verify3D [35] and by PROCHECK [36], as implemented in the PDBsum site [37]. Finally, a geometry regularization of each chimera was performed similarly to that carried out in the natural (non-chimeric) complexes.
Solvation, ionization state assignments and molecular dynamics simulations
Solvation of the complexes was performed using the Water Soak subroutine of the MOE program using a water-molecules sphere of radius 0.7 nm from protein atoms. Since the reported experimental inhibition constants were determined at pH 5.5, we adjusted the ionization state of the corresponding residues with the Protonate 3D function implemented in MOE. Both thiolate and imidazolium groups were created for the Cys and His side chains of the catalytic residues, because this is required for a fully active enzyme in vivo. Finally, once again, an energy minimization was performed until the force gradient was below 40 kJ/(mol·nm), a condition necessary to keep the water molecules from slipping away from the protein surface during the molecular dynamics (MD) calculations. Once minimized, and to allow the free movement of the atoms and give rise to reach better enzyme-prosegment fits each system was subjected to a 300 K MD simulation with MOE using a canonical ensemble (NVT), until equilibration of all systems was attained, as judged by stabilization of the RMSD values with fluctuations less than 0.001 nm.
Correlation with experimental values and interface interactions
After MD simulations the 13-amino acid-long prosegment fragment, that spans the entire active site cleft and catalytic subsites, was analyze for the last 50 conformers to evaluate two structural parameters that could correlate with the reported Ki values, [21,22]; the differential van der Waals (vdw) and electrostatic energies upon complex formation. Both parameters were calculated as ∆E = E complex - (E enzyme + E prosegment) using the Potential Energy application of MOE for the nine complexes. The best correlation was obtained for the electrostatic interface energy (∆Eelec) using a 13-residue segment. The corresponding sequences are: VVQKMTGLKVPLS (residues 71 to 83) for pCK, FRQVMNGFQNRKP (residues 71 to 83) for pCL and VMSLMSSLRVPSQ (residues 73 to 85) for pCS. To determine its specific interactions with the enzyme, each residue from these segments was analyzed with the MOE Protein Contacts tool. All hydrogen bonds and ionic prosegment-cathepsin interactions were identified, along with their corresponding contact residues in the enzyme moiety. Parameters used in this task were as follows: disulfide bond cutoff 0.25 nm, sequence separation 4, and network separation 0. Both the hydrophobic and ionic cutoffs were set to 0.65 nm.
Analysis of specific inhibitor molecules
Structures of inhibitor-cathepsin complexes available in the PDB were compared to the enzyme-ligand interactions found in this work. A 4-(methyl sulfonyl) phenyl trifluoroethylamine amide derivative (PDB code 1vsn, an analog of cathepsin K inhibitor odanacatib) in complex with cathepsin K [38]; 4-biphenylacetyl-Cys-(D) Arg-Tyr-N-(2-phenylethyl) amide (PDB code 1mhw) in complex with cathepsin L [39], and thioether acetamide P3 inhibitor (PDB code 3kwn) bound to cathepsin S [40] were used. Negative charges of catalytic thiols in the 3kwn and 1mhw files were imposed; this last step was not necessary for 1vsn, since in this case the ligand is covalently linked to the active-site cysteine of cathepsin K. The geometry of these three structures was optimized in the same way as for the natural and chimeric complexes. To illustrate the interactions established for every cathepsin inhibitor with their respective enzyme, two-dimensional diagrams were drawn with the Ligand Interactions MOE tool. These diagrams are shown in Figs. SI2 1vsn, 1mhw and 3kwn (supporting information section) for cathepsin K, L and S inhibition complexes, respectively.
Results
As mentioned in the Materials and Methods section, two intermolecular parameters were calculated for the nine modeled complexes of the human cathepsin-prosegment systems. We tested the correlation of both parameters with the Ki values and the results, using the 13-amino acid proregion fragments, are shown in Figs. 1a and 1b.
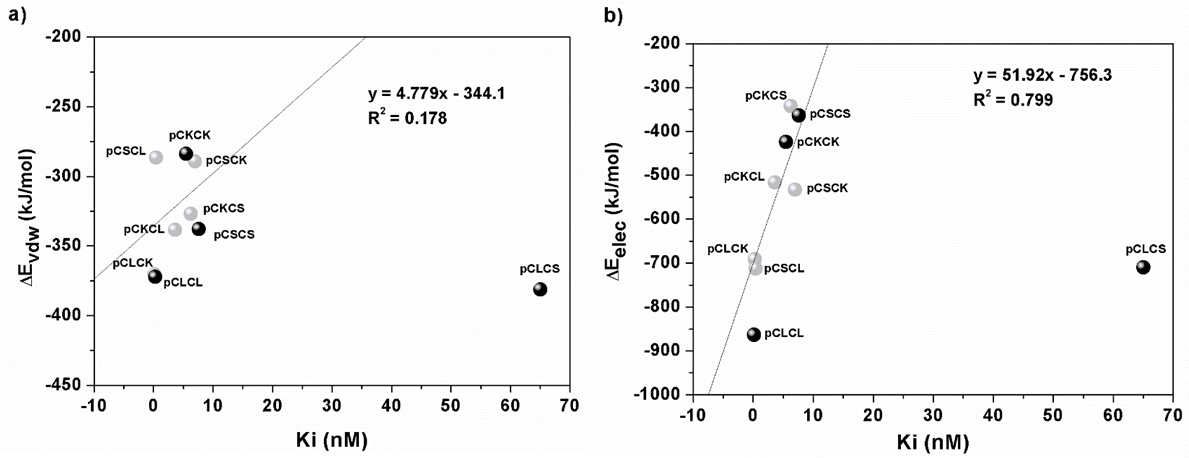
Fig. 1
Linear regression of the optimized linear correlation between Ki exp and structural ∆Evdw and ∆Eelec values, (a) and (b) respectively, as calculated with the MOE program (without considering pCLCS chimera data).
The strongest correlation with Ki values was found for the electrostatic energy of the prosegment-enzyme interaction (Fig. 1b). Interestingly, the electrostatic energy behavior in natural and chimeric complexes followed the same tendency, and the most outstanding fact is the location of the pCLCS chimera point out of the linearity. R2 values for the whole set of complexes, including this chimera, for the vdw and electrostatic interfacial energy fits to the Ki values are 0.1789 and 0.0249, respectively. An inconvenient interaction between two positively charged side chains was detected when analyzing the three-dimensional structure of pCLCS chimera; the Arg 11 from the prosegment and the Lys 64 from the enzyme. Calculus of free energies using the APBS program [41] for this chimera yielded unfavorable Coulomb interactions combined with one of the highest solvation values (Table SI1). These facts could lead to a net decrease in electrostatic binding affinity, but certainly the effect was not enough for explaining the high experimental Ki value of this chimera. We intended then to improve its three-dimensional structure by submitting the 13-amino acid prosegment fragment structure to docking simulations over the enzyme, however ΔEelec values and conformations of the best poses remained close to the initial data. Since the model of the pCLCS chimera may not be representative of the actual prosegment-enzyme binding, this structure was not considered in the following analysis.
The 13-amino acid segment used for electrostatic calculations extends from the unprimed to primed S subsites, and areas, of the active site, and it is presented with consecutive numbering in Table 1. A three-dimensional representation of the fragment on the active site crevice is shown in Fig. 2 for the pCLCL natural complex. The rest of the structures are shown in the supporting information section (Figure SI1, panels A, B and C).
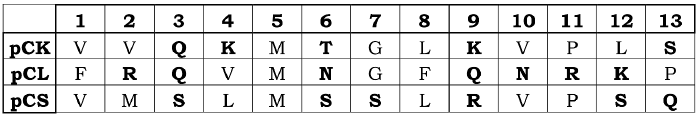
Table 1
Sequence and alignment of the 13-amino acid segments used for the electrostatic interaction calculations of prosegment-enzyme complexes, and also for the electrostatic profiles. Polar and positively charged side chain residues are highlighted in bold type.
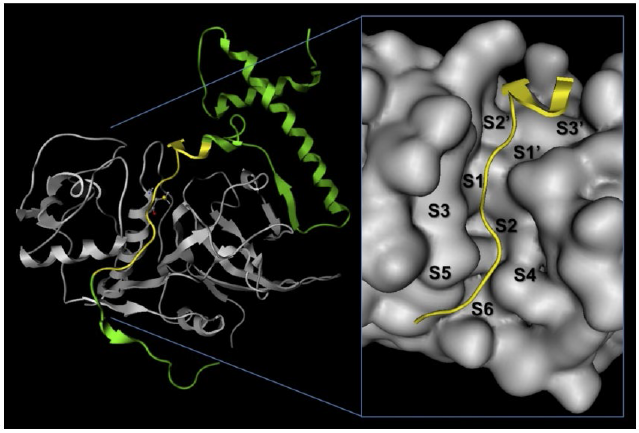
Fig. 2
Left: Ribbon representation of the pCLCL complex with the prosegment (shown in green) and the 13-amino acid interaction segment (in yellow). Right: Surface of the active site crevice of the mature enzyme. The catalytic subsites and areas are marked with letters and numbers. The yellow ribbon represents the 13-amino acid segment used for electrostatic calculations.
Analysis of natural complexes
To describe the nature of the prosegment-enzyme interaction in more detail, the electrostatic binding energy was calculated for each one of the 13 residues of the segments. The resulting highest-energy values are shown in Fig. 3.
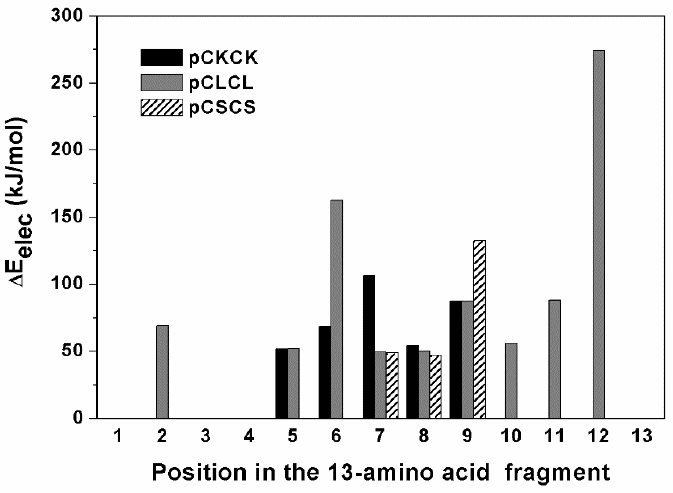
Fig. 3
Residues of the 13-amino acid fragments interacting with their correspondent mature enzyme. Numbering refer totothose positions in Table 1 for all three peosegment chains studied. To emphasize the most important contacts, only the interactions with energies equal to, or higher than, 40 kJ/mol are shown.
Superposition of the entire prosegments upon binding their cognate enzymes yields an angle of approximately 30° between their third helix, with an RMSDCα value of 2.31 for the pCS and pCL proregions (Fig. 4). These results could explain the differences in selectivity of the enzyme for its prosegment during intermolecular recognition.
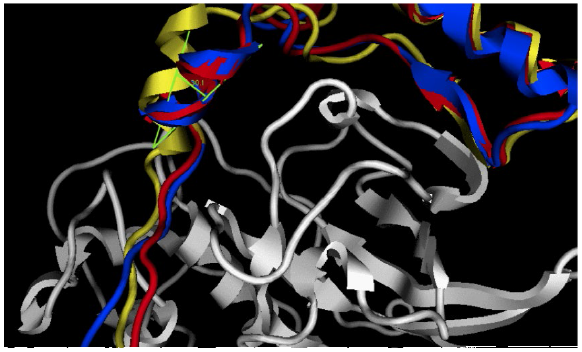
Fig. 4
Ribbon representation of the crystallographic structures for all three natural complexes of human cathepsin L (red), K (blue) and S (yellow) over the overlapped structures of their mature enzymes (RMSD Cα = 0.112 nm) after MD simulation. The angle between the third prosegment helixes of cathepsin S (pCS) and L (pCL) is shown in green. As a reference, the mature moiety of cathepsin L (CL) is shown in white ribbons.
Comparison between interface interactions in natural and chimeric complexes
To make a comparative analysis of the chimeras and natural complexes, the differential electrostatic energies of the interfacial contacts were calculated over the 13-amino acid segments for the chimeric complexes and their corresponding natural ones. The results are shown in Fig. 5 and Table SI2.
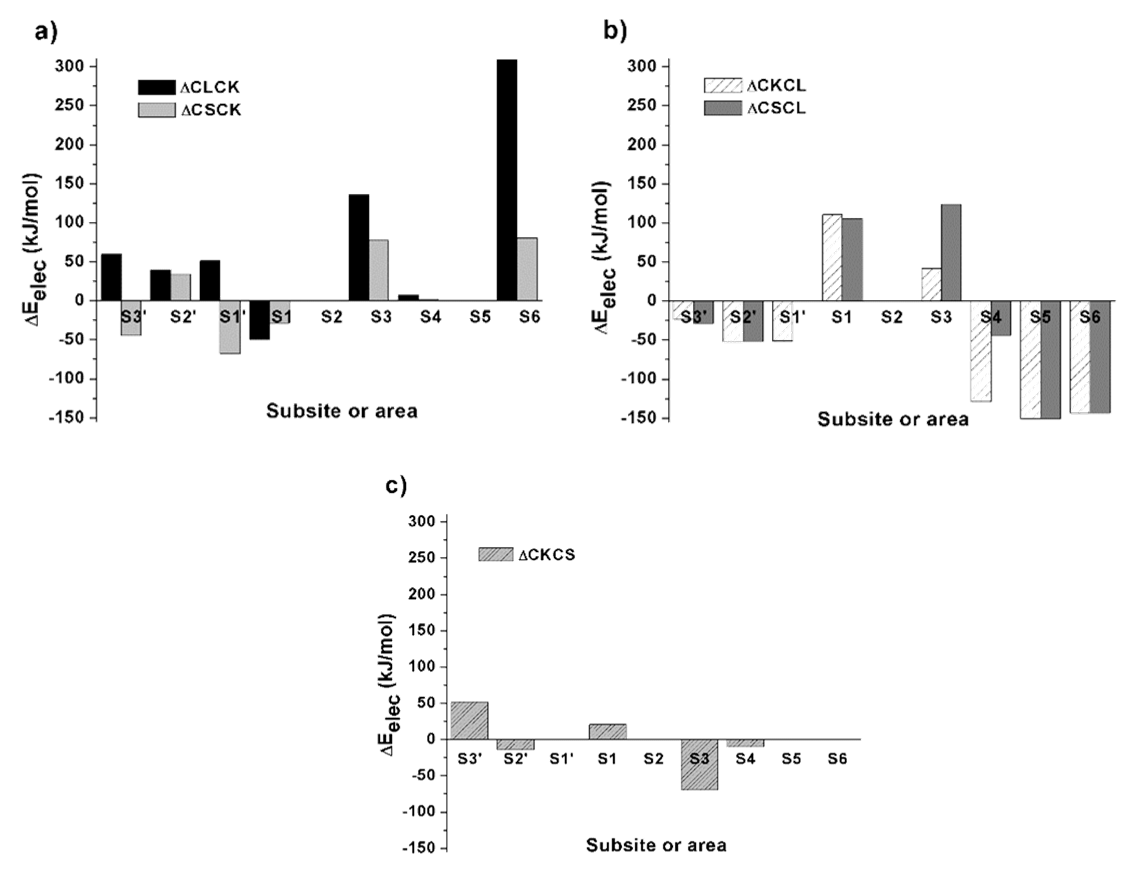
Fig. 5
Electrostatic energy profiles generated by subtracting the value of each identified interaction in the chimeric complexes from the interactions of the natural complexes for the same catalytic subsites and areas, in the active site cleft. Positive values indicate more favorable interactions in the chimera than in the natural complex. a), b) and c) show differential energetic profiles of the chimeric complexes of CK, CL and CS with respect to their natural complexes.
To visualize details of every enzyme-prosegment interaction in the natural complexes and to identify the contributions of chimeric contacts, we made the diagram of contacts shown in Figure 6. For comparison purposes, this figure also includes the interactions seen between enzyme moieties and the specific cathepsin inhibitors analyzed in this work.
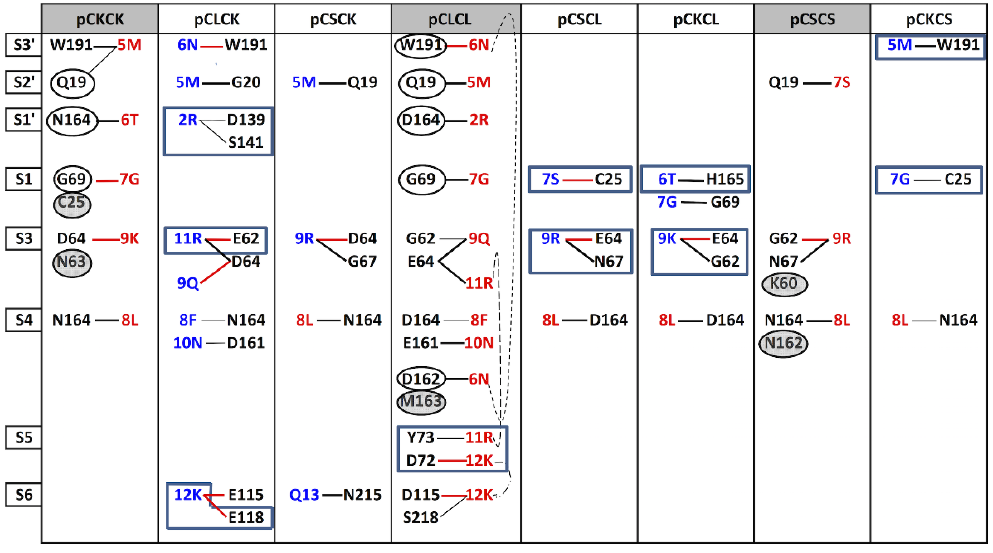
Fig. 6
Diagram of contacts for the enzymatic subsites and zones observed in the enzyme-prosegment interface complexes. Grey cells indicate natural complexes. The prosegment numbering is inverted. Red and black symbols indicate proregion and enzyme residues, respectively. Blue symbols indicate better contacts in chimeras than in natural complexes. Thick lines represent strong interaction energies (black, greater than 40 kJ/mol; red, greater than 80 kJ/mol). Ovals indicate contacts of specific inhibitors upon binding to the enzyme moiety; open symbols indicate common interactions with prosegments; closed ovals indicate interactions absent in prosegment-enzyme complex. Boxes indicate potential or current residues to be considered for the design of potent inhibitors. Segmented lines show the presence of the same proregion residue in two different subsites.
Discussion
Natural complexes
Our results indicate that the highly energetic electrostatic interactions established by the proregions are more widely distributed and more highly interconnected in pCLCL than in the other two enzymes (Fig. 3 and 6). These facts could explain the significantly lower inhibition constant value for this complex (Ki = 0.12 nM). Moreover, the S5 and S6 areas (particularly the latter) are important in the interface of the pCLCL complex because they establish important intermolecular ionic-pair interactions.
S2 subsite, not shown in Fig. 6, establishes hydrophobic interactions with the side chains of the eighth residues of the 13-amino acid prosegment fragments and it is the main chain of these residues which actually interact electrostatically with S4 subsite.
Comparative analysis of the three enzymes shows several coincidences upon binding to their cognate proregion, as shown in Fig. 3 and Table 1. For example, the seventh position includes a small amino-acid (7G or 7S) whilst position number eight is always hydrophobic (8L and 8F). Also, position number nine in Table 1 shows a large or basic amino acid group. These ninth and eighth residues show interactions with the enzymatic portion in the three natural complexes with different residues of the S3 and S4 subsites, respectively (Fig. 6). On the other hand, the seventh residue interacts with the S1 subsite in pCKCK and pCLCL, and with the nearby subsite S2' in pCSCS (Figs 6, and SI1, panels A, B, C, respectively).
The greatest differences in the enzyme-prosegment electrostatic binding of the pCLCL complex compared to those interactions in the other two natural complexes are due to five residues (the second, the sixth and from the tenth to the twelfth, Fig. 3), which bind to S1', S3, S4, S5 and S6 sites and areas, respectively (Fig. 6). The last three zones are located at the lower end of the active site cleft (Fig. 2). Those five subsites and areas probably determine the specificity of the intermolecular recognition in the pCLCL complex. Furthermore, residues Tyr 73 and Asp 72 of the S5 area of CL bind to positively charged residues of the cognate prosegment and do not have any similarities with the rest of the natural complexes studied, therefore, they could be employed to improve the specificity of binding to this enzyme for inhibitor design. These residues and their contacts with the prosegment are marked with a box in Fig. 6 for the pCLCL complex.
The highest energy electrostatic interactions between the enzymes and their cognate proregions are in the S3' and S3 subsites of the pCKCK complex; the S3', S5 and S6 subsites of the pCLCL complex, and the S3 subsite of pCSCS (Figure 6). All of these zones are of great interest as potent inhibition sites of the three cathepsins studied in this work.
Chimeric complexes
The great number of electrostatic interactions, established by pCL attached to CK, is remarkable when compared to those formed by the pCK on its cognate enzyme in the natural complex (pCKCK in Fig. 6). Indeed, the binding energy in the chimeric complex is significantly improved, as judged by the five interactions of positive bars in Fig. 5a, highlighted with wide red lines in Fig 6. for the pCLCK chimera. Several of these interactions are similar to those of the natural complex pCLCL, which is in agreement with their respective inhibition constant values that are the lowest of all the complexes studied in this work (Ki = 0.27 nM and 0.12 nM). Fig. 6 shows the great affinity for the same proregion in these two complexes and reveals the difficulty in designing specific inhibitors for CK and CL human proteases. Beyond these similarities, there are some small differences that could be used for specific binding to cathepsin K, revealed by the pCLCK chimera. For example, residues Glu 62 of the S3 subsite, Asp 139 and Ser 141 of the S1' subsite and Glu 118 of the S6 area are not involved in pCL binding to its cognate enzyme. Perhaps they could be used for the design of specific inhibitors of CK, with respect to CL (because CS could also be inhibited in the S3 subsite). The aforementioned residues are marked with boxes for the pCLCK chimera in Fig 6. and, according to Fig. 5a, they correspond to the same enhanced sites on pCSCK chimera.
The pCSCL and pCKCL chimeras improve electrostatic binding to the CL enzyme only in the S1 and S3 subsites (Fig. 5b), which present polar residues Ser and Thr (7S and 6T) and positively charged amino acids Arg and Lys (9R and 9K) at pH 5.5, instead of Gly and Gln (7G and 9G) in the natural proregion according to Fig. 6. Despite these better contacts, the inhibition constants of both chimeras (0.46 nM and 3.6 nM for pCSCL and pCKCL, respectively) are far from the Ki value of the natural complex (0.12 nM). This could be due to the absence of the most favorable interactions in the S5 and S6 areas of those chimeras, as well as the lack of the interconnections present in the pCLCL natural complex. Because these chimeric interactions represent improvements in the binding of the natural complex, they are marked with boxes in Fig. 6 for pCSCL and pCKCL.
Finally, the chimeric complex pCKCS (Ki = 6.3 nM) has two electrostatic interactions that improve the binding in the natural pCSCS complex (Ki = 7.6 nM). These interactions involve the fifth and seventh residues of the pCK prosegment, which bind to the S3' and S1 subsites of the CS enzyme, respectively; with the first contact being more intense than the second (Fig. 5c and Fig. 6). Because these interactions may be used in the development of specific inhibitors of CS, they are marked with boxes for the pCKCS chimera in Fig. 6.
Small inhibitors in all three cathepsins
Analysis of the crystallographic structures of cathepsins K, L and S with specific organic inhibitors from the PDB (codes 1vsn, 1mhw and 3kwn, respectively) indicates that most of the interactions employed by them (8 out of 13) coincide with the sites identified for the prosegments in the natural complexes, as shown by open ovals in Fig. 6. Participating residues in all these interactions are shown on Figs. SI2. Moreover, the rest of the zones explored by the inhibitors correspond to the same sites observed in the prosegments; this is the case for the S1 and S3 subsites in pCKCK (enzyme residues Cys 25 and Asn 63 in Fig. SI2, 1vsn), the S4 subsite in pCLCL (enzyme residue Met 163, Fig. SI2, 1mhw) and the S3 and S4 subsites in pCSCS (enzyme residues Lys 60 and Asn 162, Fig. SI2, 3kwn). All these interactions are also indicated with closed ovals in Fig. 6. As additional data, it is possible to identify the participation of hydrophobic groups reinforcing electrostatic contacts in cathepsin S inhibition (Fig. SI2, 3kwn). The coincidences between the binding to enzymes of these small specific inhibitors, and those of the proregion fragments, support our findings regarding the way prosegments interact with enzymes active site cleft in the modelled complexes generated in the present work.
Other reported facts on cathepsins inhibition
The parameter selected in this work for the study of intermolecular interactions is in agreement with the experimental fact reported by Löser and Pietzsch [9] on that, most of the known low molecular weight inhibitors of papain-like proteases contain electrophilic entities.
The high selectivity of the cathepsin S prosegment (pCS) for CL, and even for CK, reported by Guay et al. [3] can be explained, by our results, through the establishment of additional-to-natural interactions in the S1 subsite of CL and in the S6 area of CK.
Results published by Kramer et al. [10] on the substrate specificity of the same three cathepsins studied here correspond to the physicochemical properties observed for their natural proregions in S2, S3 and S4 subsites, except that, for these last two sites, prosegment inhibition is better performed by positive charged residues.
The 13-amino acid fragments studied here bind to primed enzyme regions equivalent to those observed for the first beta-hairpin loop and the N-terminal segment of the stefin B inhibitor in the active site cleft of papain [20], a close homolog to cathepsins. This fact may indicate the existence of some molecular patterns common to cysteine protease inhibitors.
The lack of inhibition power of the MNGFQ pentapeptide against cathepsin L, reported by Chowdhury et al. [39], can be explained by our results considering that this pentapeptide is missing the most important residues for the molecular recognition of S1’and from the S3 to S6 subsites and areas, i.e., Arg 2, Asn 10, Arg 11, and Lys 12 (2R, 10N, 11R and 12K using prosegment numbering of Table 1 and Fig. 6).
A lower inhibition power for an 81-amino acid fragment of the human cathepsin L prosegment compared with the quasi-complete prosegment chain has been reported by Carmona et al. [42]. This result can be explained by the lack of the Lys 12 residue in the proregion, which strongly attaches to the S5 and S6 areas of the enzyme (12K in Fig. 6). On the other hand, we hypothesize that the failure of the C-terminal 44-amino acid-long proregion to properly inhibit human cathepsin L, presented in the same work [42], might not be due to the absence of adequate residues for recognition, because it has the entire 13 residues of the proregion fragment, but rather to the lack of secondary structural elements (such as the first and second prosegment helixes), which could alter the conformational stability of the prosegment itself. This explanation is plausible because a destabilizing conformational change was proposed by Cappetta et al. [43] when they introduced mutations (far from the active site) in the GNFD motif of the Fasciola hepatica cathepsin L prosegment and it was detected only marginal enzymatic activity. Thus, the conformational effects of the entire propeptide moieties on the proteolytic activity control still requires more detailed studies.
Conclusions
Comparison of the interfacial electrostatic interactions in natural complexes indicate that five sites and areas, S1' and the S3, S4, S5 and S6, probably determine the specificity of the intermolecular recognition in the pCLCL complex. Furthermore, the highest energy bonds are found in the S3' and S3 subsites for the cathepsin K complex (pCKCK); S3', S5 and S6 for the cathepsin L complex (pCLCL), and S3 for the cathepsin S complex (pCSCS). Among them, cathepsin L establishes the highest number of both electrostatic interactions and interconnections with its natural proregion, revealing the S5 and S6 areas to be high-energy anchoring sites. Moreover, residues Asp 72 and Tyr 73 in the S5 area of cathepsin L can increase the selectivity of the enzyme to charged and polar groups of inhibitor molecules.
Regarding chimeric complexes, pCLCK reveals the great similarity between the receptor residues in the catalytic subsites of cathepsins K and L. This fact could hinder the design of specific inhibitors through electrostatic interactions for these two enzymes. Furthermore, chimeric complexes pCSCL and pCKCL improve the electrostatic binding to the enzyme with respect to the natural complex pCLCL only in subsites S1 and S3, because the proregions in these chimeric complexes present polar and positively charged residues. The pCKCS chimeric complex has two electrostatic interactions that improve natural binding to the enzyme at the S3' and S1 subsites. Proregion groups involved here are the polar side chain of a methionine (5M) and the main chain of a glycine (7G).
In this study, electrostatic interactions proved to be a reliable parameter to describe the intermolecular prosegment-enzyme interface of the analyzed cathepsin complexes. Furthermore, experimental results from other research studies on the inhibition of cathepsins could be explained at a molecular level, such as the cathepsin L inhibition by fragments of its own prosegment or peptidomimetic compounds, the selectivity of cathepsin S prosegment for cathepsins L and K, and the common zones used by prosegments and other ligand proteins to inhibit papain and the cathepsin enzymes studied here. This last fact suggests that the study of electrostatic interactions may be useful to elucidate relevant interactions in other members of the cysteine protease family.
Acknolwedgements
JCM-H was supported by the National Council of Science and Technology (CONACYT, Mexico) [scholarship number: 194059].
References
1. Schick, C.; Pemberton, P.A.; Shi, G.P.; Kamachi, Y.; Cataltepe, S.; Bartuski, A.J.; Gornstein, E.R.; Bromme, D.; Chapman, H.A.; Silverman, G.A. Biochem. 1998, 37, 5258-5266.
2. Palermo, C.; Joyce. J.A. Trends Pharmacol Sci. 2007, 29, 22-28.
3. Schornberg, K.; Matsuyama, S.; Kabsch, K.; Delos, S.; Bouton, A.; White, J. J. Virol. 2006, 80, 4174-4178.
4. Lankelma, J.M.; Voorend, D.M.; Barwari, T.; Koetsveld, J.; Van der Spek, A.H.; De Porto, A.P.N.A.; Van Rooijen, G.; Van Noorden, C.J.F. Life Sciences. 2010, 86, 225-233.
5. Bromme, D.; Klaus, J.L.; Okamoto, K.; Rasnick, D.; Palmer, J.T. Biochem. J. 1996, 315, 85-89.
6. Cywin, C.L.; Firestone, R.A.; McNeil, D.W.; Grygon, C.A.; Crane, K.M.; White, D.M.; Kinkade, P.R.; Hopkins, J.L.; Davidson, W.; Labadia, M.E.; Wildeson, J.; Morelock, M.M.; Peterson, J.D.; Raymond, E.L.; Brownand, M.L.; Spero, D.M. Bioorg. Med. Chem. 2003, 11, 733-740.
7. Gauthier, J.Y.; Chauret, N.; Cromlish, W.; Desmarais, S.; Duong, L.T.; Falgueyret, J.P.; Kimmel, D.B.; Lamontagne, S.; Le´ger, S., Le Riche, T.; Sing, C.; Masse´, L.F.; McKay, D.J.; Nicoll-Griffith, D.A.; Oballa, R.M.; Palmer, J.T.; Percival, M.D.; Riendeau, D.; Robichaud, J.; Rodan, G.A.; Rodan, S.B., Seto, C.; The´rien, M.; Truong, V.L.; Venuti, M.C.; Wesolowski, G.; Young, R.N.; Zambonia, R.; Black, W.C. Bioorg. Med. Chem. Lett. 2008, 18, 923-928.
8. Stoch, S.A.; Wagner, J.A. Clin. Pharmacol. Ther. 2008, 83, 172-176.
9. Löser, R.; Pietzsch, J. Cysteine cathepsins. Front Chem. 2015, 23, 3-37.
10. Kramer, L.; Turk, D.; Turk, B. Trends Pharmacol Sci. 2017, 38, 873-889.
11. Coulombe, R.; Grochulski, P.; Sivaraman, J.; Ménard, R.; Morton, J.S. Cygler, M. EMBO J. 1996, 15, 5492-5503.
12. Maubach, G.; Schilling, K.; Rommerskirch, W.; Wenz, I.; Schultz, J.E.; Weber, E.; Wiederanders, B. Eur. J. Biochem. 1997, 250, 745-750.
13. Billington, C.J.; Mason, P.; Magny, M.C.; Mort, J.S. Biochem. Biophys. Res. Commun. 2000, 276, 924-929.
14. Nomura, T.; Fujisawa, Y. Biochem. Biophys. Res. Commun. 1997, 230, 143-146.
15. Ishidoh, K.; Saido, T.C.; Kawashima, S.; Hirose, M.; Watanabe, S.; Sato, N.; Kominami, E. Biochem. Biophys. Res. Commun. 1998, 252, 202-207.
16. Ménard, R.; Carmona, E.; Takebe, S.; Dufour, E.; Plouffe, C.; Mason, P.; Mort, J.S. J. Biol. Chem. 1998, 273, 4478-4484.
17. Rieman, D.J.; McClung, H.A.; Dodds, R.A.; Hwang, S.M.; Holmes, M.W.; James, I.E.; Drake, F.H.; Gowen, M. Bone. 2001, 28, 282-289.
18. Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. Nucleic Acids Res. 2000, 28, 235-242.
19. Schechter, I.; Berger, A. Biochem. Biophys. Res. Commun. 1967, 27, 157-162.
20. Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Biochim. Biophys. Acta. 2012, 1824, 68-88.
21. Guay, J.; Falgueyret, J.P.; Ducret, A.; Percival, M.D.; Mancini, J.A. Eur. J. Biochim. 2000, 267, 6311-6318.
22. Wiederanders, B. Acta. Biochim. Pol. 2003, 50, 691.
23. Ang, K.K.H.; Ratnam, J.; Gut, J.; Legac, J.; Hansell, E.; Mackey, Z.B.; Skrzypczynska, K.M.; Debnath, A.; Engel, J.C.; Rosenthal, P.J.; McKerrow, J.H.; Arkin, M.R. and Renslo, A.R. PLoSNegl. Trop. Dis. 2011, 5, e1023, doi:10.1371/journal.pntd.0001023.
24. Sosic, I.; Mirkovic, B.; Arenz, K.; Stefane, B.; Kos, J. and Gobec, S. J. Med. Chem. 2012, 56, 521-533.
25. Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2018.
26. Gasteiger, J.; Marsili, M. Tetrahedron. 1980, 36, 3219-3228.
27. Londe, J.M.L.; Zhao, B.; Janson, C.A.; D'Alessio, K.J.; McQueney, M.S.; Orsini, M.J.; Debouck, C.M.; Smith, W.W. Biochem. 1999, 19. 862-869.
28. Cygler, M. ; Coulombe, R. Crystal structure of procathepsin L doi: 102210/pdb1CS8/pdb.
29. Kaulmann, G.; Palm, G.J.; Schilling, K.; Hilgenfeld, R.; Wiederanders, B. Protein Sci. 2006, 15; 2619-2629.
30. Nucleic Acids Research, Volume 45, Issue D1, 4 January 2017, Pages D158-D169, https://doi.org/10.1093/nar/gkw1099.
31. Inaoka, T.; Bilbe, G.; Ishibashi, O.; Tezuka, K.; Kumegawa, M.; Kokubo, T. Biochem. Biophys. Res. Commun. 1995, 206, 89-96.
32. Gal, S.; Gottesman, M.M. Biochem. J. 1988, 253, 303-306.
33. Shi, G.P. Munger, J.S.; Meara, J.P.; Rich, D.H.; Chapman, H.A. J. Biol. Chem. 1992, 267, 7258-7262.
34. Söding, J.; Biegert, A.; Lupas, A.N. Nucleic Acids Res. 2005, 33, 244-248.
35. Eisenberg, D.; Lüthy, R.; Bowie, J.U. Methods Enzymol. 1997, 277, 396-404.
36. Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. J. Appl. Cryst. 1993, 26, 283-291.
37. Laskowski, R. A. Nucleic Acids Res. 2009, 37, D355-D359.
38. Li, C.S.; Deschenes, D.; Desmarais, S.; Falgueyret, J.P.; Gauthier, J.Y.; Kimmel, D.B.; Léger, S.; Massé, F.; McGrath, M.E.; McKay, D.J.; Percival, M.D.; Riendeau, D.; Rodan, S.B.; Thérien, M.; Truong, V.L.; Wesolowski, G.; Zamboni, R.; Black, W.C. Bioorg. Med. Chem. Lett. 2006, 16, 1985-1989.
39. Chowdhury, S.F.; Sivaraman, J.; Wang, J.; Devanathan, G.; Lachance, P.; Qi, H.; Ménard, R.; Lefebvre, J.; Konishi, Y.; Cygler, M. ; Sulea, T.; Purisima, E.O. J. Med. Chem. 2002, 45, 5321-5329.
40. Wiener, D.K.; Lee-Dutra, A.; Bembenek, S.; Nguyen, S.; Thurmond, R.L.; Sun, S.; Karlsson, L.; Grice, C.A.; Jones, T.K.; Edwards, J.P. Bioorg. Med. Chem. Lett. 2010, 20, 2379-2382.
41. Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Proc. Natl. Acad. Sci. 2001, 98, 10037-10041.
42. Carmona, E.; Dufour, E.; Plouffe, C.; Takebe, S.; Mason, P.; Mort, J.S.; Ménard, R. Biochem. 1996, 35,8149-8157.
43. Cappetta, M.; Roth, I.; Díaz, A.; Tort, J.; Roche, L. Biol. Chem. 2002, 383, 1215-122.
Author notes
*Corresponding author: Jaqueline Padilla-Zúñiga, e-mail: jpz@xanum.uam.mx, Universidad Autónoma Metropolitana-Iztapalapa, Departamento de Química, San Rafael Atlixco 186, Col. Vicentina, 09340 Iztapalapa, Mexico City, Mexico.
