Journal of the Mexican Chemical Society, vol. 61, no. 3, 2017
Sociedad Química de México A.C.
Oscar Palomino-Hernández
Universidad Nacional Autónoma de México, Mexico
A. Christiaan Jardínez-Vera
Universidad Nacional Autónoma de México, Mexico
José L. Medina-Franco *
Universidad Nacional Autónoma de México, Mexico
Date received: 06 January 2017
Date accepted: 21 April 2017
Funding
Funding source: Universidad Nacional Autónoma de México (UNAM)
Contract number: IA204016
Funding
Funding source: Facultad de Química, UNAM
Contract number: 50009163
Funding
Funding source: CONACyT
Contract number: 291053
Funding
Funding source: CONACyT
Contract number: 765897/606003
Abstract: Abstract. Inhibitors of DNA methyltransferases 3A and 3B (DNMT3A/3B) are promising candidates for the treatment of cancer and other diseases. Selective inhibitors of DNMT3A/3B are also attractive as small-molecule probes. During the past few years has increased significantly the research towards the development of DNMT1 inhibitors. However, there are no reviews of the recent progress in the development of small-molecule inhibitors of DNMT3A/B. Herein we review the status of inhibitors of DNMT3A/3B with emphasis on computational guided approaches. We discuss in a critical manner compound databases containing structure-activity information, crystallographic structures of DNMT3s, structure-guided studies, and virtual (in silico) screening coupled with experimental validation that have led to the identification or development of selective inhibitors. Perspectives in the field are also discussed.
Keywords: Drug discovery, Epi-informatics, epigenetics, DNA methyltransferase, structure-activity relationships.
Resumen: Resumen. Inhibidores de metiltransferasas de ADN 3A y 3B (DNMT3A/3B) son candidatos prometedores para el tratamiento de cáncer y otras enfermedades. Inhibidores selectivos de DNMT3A/3B también son atractivos como moléculas sonda. En los últimos años se ha incrementado el desarrollo de inhibidores de DNMT1. Sin embargo, no hay trabajos de revisión que abarquen el desarrollo de inhibidores de DNMT3A/3B. En este trabajo se revisa el estado actual de la investigación de inhibidores de DNMT3A/3B con énfasis en las estrategias guiadas por métodos computacionales. Se discuten en forma crítica bases de datos moleculares que contienen información de relaciones estructura-actividad, estructuras cristalográficas de DNMT3s, estudios guiados por la estructura y estudios de evaluación virtual unidos a caracterización experimental para la identificación de inhibidores selectivos. También se discuten perspectivas en este campo.
Palabras clave: Descubrimiento de fármacos, Epi-informática, epigenética, ADN metiltransferasa, relaciones estructura-actividad.
1. Introduction
DNA methylation is one of the many epigenetic modifications associated with genetic expression in higher eukaryotes, regulating the processes of DNA transcription and peptide synthesis due to chromatin remodelling. The covalent modification is attributed to the catalytic action of DNA methyltransferases (DNMTs) [(1)], enzymes which can either promote de novo methylation (DNMT3A and DNMT3B) or maintenance me thy lation (DNMT1), following DNA replication. To date, only these three types of DNMTs with catalytic activity have been identified in the human genome [(2)], whereas the fourth type, namely DNMT3L, is a highly homologous nuclear protein that lacks the amino acid residues required for catalytic activity.
Regular function of DNMTs enzymes are crucial for DNA methylation in both cellular development and mitosis. However, overexpression of DNMTs can lead to aberrant methylation patterns [(3)], which have been associated with many cancer types including lung, colorectal, prostate, breast, cervical and pancreatic cancer, among others [(4-6)]. Experimentally, it has been observed that DNMT inhibition reactivates silenced (hypermethylated) genes, particularly tumor-suppressor genes [(7)]. Hence, DNMTs are promising biological targets for the design of anti-cancer agents. Also, DNMTs are becoming relevant targets in the pathology of other diseases such as diabetes, obesity [(8)], Alzheimer’s disease [(9)], and other central nervous system disorders [(10)].
De novo methylation is the main activity of the DNMT3A and DNMT3B enzymes [(11)], enabling key epigenetic modifications essential for processes such as cellular differentiation and embryonic development, transcriptional regulation, heterochromatin formation, X-chromosome inactivation, imprinting, and genome stability. Albeit very similar in structure, these two enzymes appear to have different roles in mammals [(12)].
In 2015 Medina-Franco et al., reviewed inhibitors of DNMTs with emphasis on inhibitors of DNMT1. In that work, the authors discussed different in-silico studies used in the development of DNMTs. Examples of techniques discussed were molecular docking, virtual screening, pharmacophore model, molecular dynamics, and similarity searching [(13)]. In 2016 Prieto-Martínez, et al. discussed advances in computational approaches applied mostly to DNMT1 [(14)]. However, there are no reviews focused on the computational development on DNMT3A and 3B inhibitors. This is because the development of inhibitors of DNMT3A and DNMT3B has been slower. In this work, we summarize recent developments of small molecule inhibitors of DNMT3A and 3B as potential therapeutic agents or molecular probes. The review focuses on the role of computational methods that have contributed to such developments.
2. DNMTs: structure and mechanism
Mammalian DNMTs have a high level of homology, possessing a large N-terminal region of variable size and a C-terminal catalytic portion. Whereas the N-terminal region encodes regulatory functions, the C-terminal region is involved in both cofactor binding and substrate catalysis. These enzymes rely on S-adenosyl-L-methionine (AdoMet or SAM) as a methyl group donor and contain several highly conserved structural features such as the SAM binding pocket, the DNA-cytosine binding pocket, and a vicinal proline-cysteine pair important for the reaction mechanism. However, there are structural differences between DNMTs: while DNMT1 relies on its N-terminal domain [(15)] to recognize hemimethylated strands (hence its function in maintaining the methylation status), DNMT3A and DNMT3B do not show any preference about the methylation status of the DNA, rendering them as de novo transferases. The 3D coordinates of DNMTs with different domains have already been deposited in the Protein Data Bank (PDB) [(16)]. Based on the structural information, mostly obtained from X-ray crystallography, it is plausible to consider at least three types of inhibitors, namely; (a) molecules binding in the co-factor binding pocket (allosteric inhibition), (b) molecules binding in the DNA binding pocket (competitive inhibition), and (c) molecules interacting directly with the catalytic cysteine (irreversible inhibition). Fig. 1 shows the catalytic domains of DNMT3A and DNMT3B. For illustration purposes, this figure shows the DNA-binding domain of human and bacteria DNMT3A and 3B, respectively.
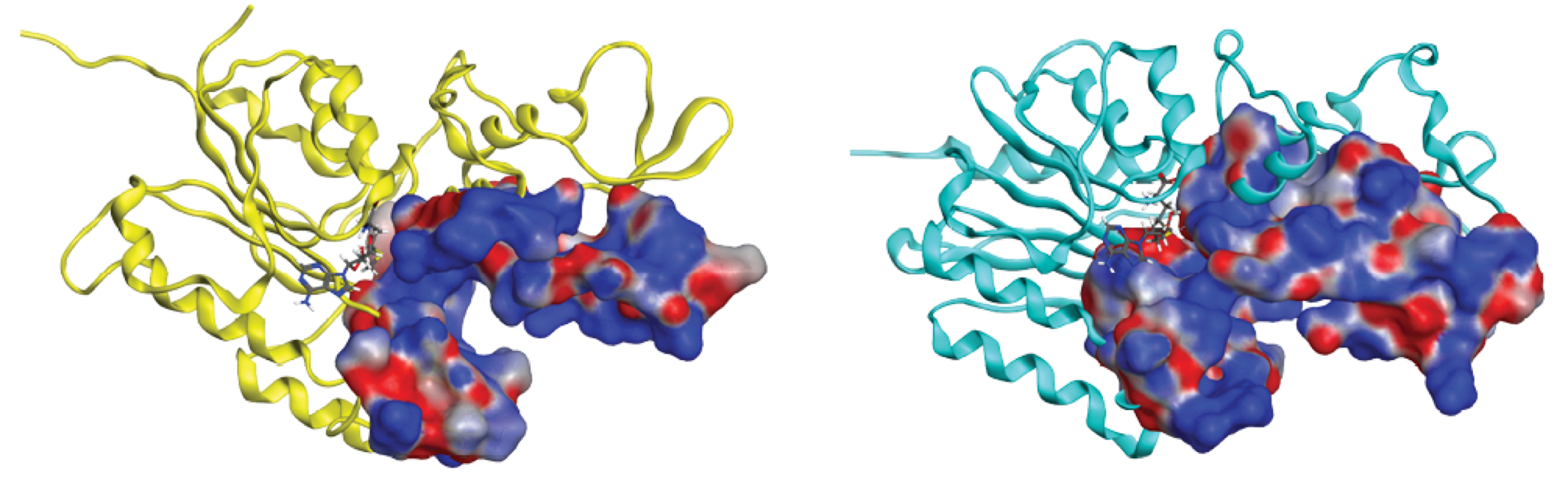
Fig. 1
Catalytic domains of DNMT3A (yellow, PDB ID: 2QRV) and DNMT3B (blue, PDB ID: 5CIY). The electrostatic molecular surface shows the DNA-binding domain. The reduced cofactor SAH is shown in both structures.
3. Modulators of DNMT3A and DNMT3B
In order to explore the chemical space of the current inhibitors a molecular database of inhibitors of DNMT3A and DNMT3B was assembled following a previously published protocol [(17)]. Six public databases and two sources of scientific literature were explored. The public databases explored were (1) ChEMBL [(18)], (2) Therapeutic Target Database (TTD) [(19)], and (3) ChEpiMod [(20)] using the query text ’DNMT3A’ or ’DNMT3B’; (4) HEMD [(21)] with the information located in the enzyme browser, option DNA and submenu DNA (cytosine-5)-methyltransferase 3A or 3B; (5) Binding Database [(22)] searching in the IC50 menu, submenu DNA methyltransferase A or B; and (6) ChromoHub [(23)] clicking on DNMT, option ‘browse inhibitors’. To retrieve additional compounds not reported in those public databases, we also reviewed the scientific literature using Web of Science (webofscience.com) and Chemical Abstracts (scifinder.cas.org). After data curation, 269 unique structures with reported activity were identified. It was found that ChEMBL, HEMD and Chemical Abstracts contained the information provided by the other chemical databases, therefore only these three sources are shown in Table 1. The analysis of the database allowed the identification of two classifications of inhibitors: (a) nucleosidic or (b) non-nucleosidic. Fig. 2 shows representative chemical structures of molecules that have been proposed as nucleoside and non-nucleoside inhibitors.
Sources to build the database of inhibitors of DNMT3.

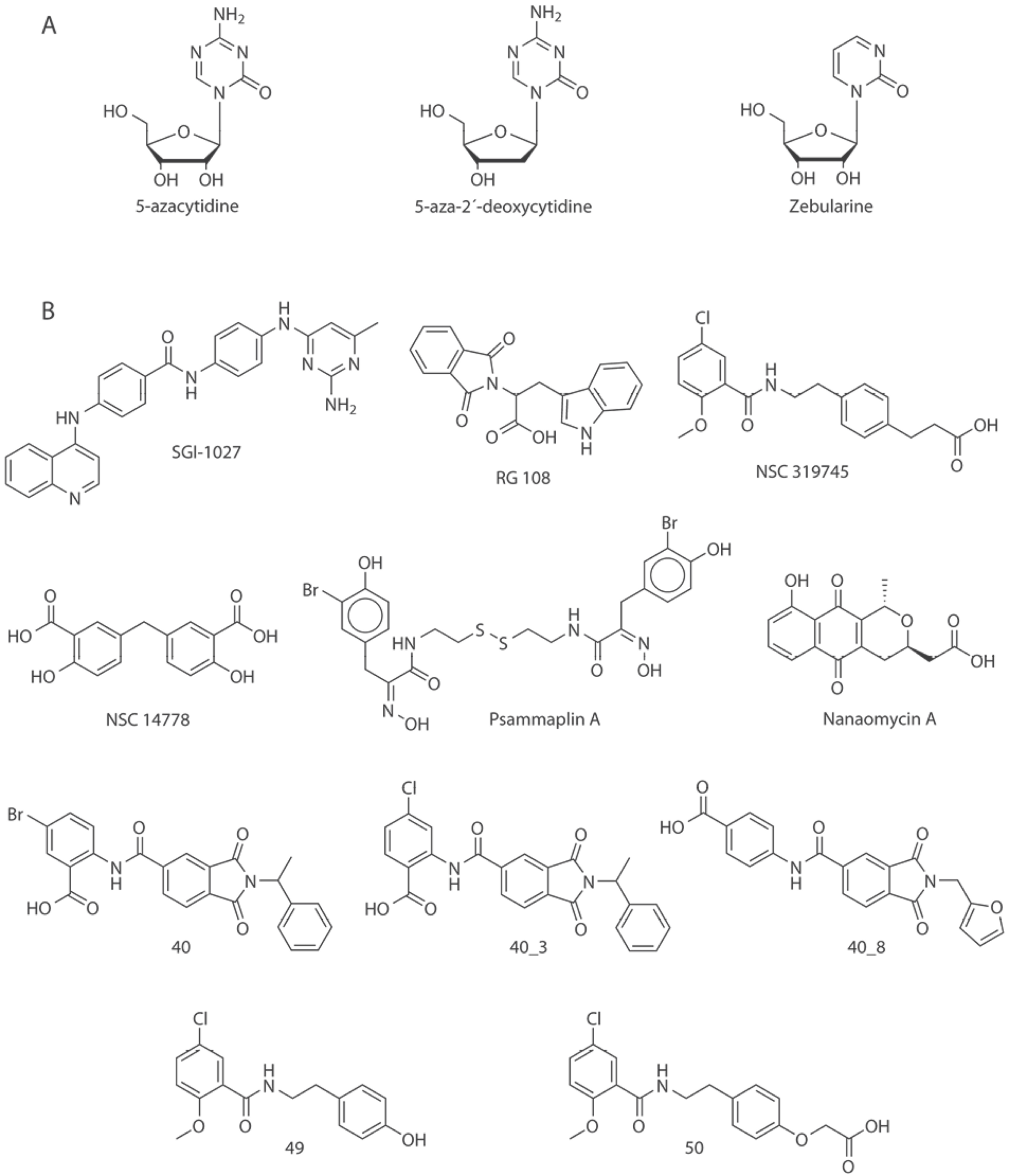
Fig. 2
Chemical structures of selected DNMT3 inhibitors (DNMTi) and other compounds associated with hypomethylating properties. Representative compounds in this figure are roughly classified as A) nucleosidic and B) non-nucleosidic inhibitors.
Chemoinformatic tools enable the management and mining of the chemical information in compound databases [(24)]. Computer-assisted molecular design methods have been used to calculate the distribution of molecular properties of inhibitors of DNMT1 [(17,25)]. Therefore, it is warranted the chemoinformatic analysis of modulators of DNMT3A/3B. These analyses contribute to the ongoing effort of charting the epigenetic-relevant chemical space (ERCS) [(25)].
4. Role of computational methods towards the development of DNMTs
Computational methods have become a corner stone in the development of bioactive compounds [(26)]. The in silico development of inhibitors of DNMT is not exception as demonstrated by the emerging ‘Epi-informatics’ field [(27)]. Computer-aided approaches can be divided in two major groups depending on the experimental information used. Structure-based approaches rely on the availability of three-dimensional information of the molecular target, whereas the ligand-based approaches depend mostly on the structure-activity information of small molecule modulators. In this section, we discuss the progress on computational studies towards the development of DNMT3A/3B inhibitors. Several methods can be classified as structure-based methods but representative ligand-based approaches have been developed as well.
4.1 Structure-based analysis of modulators of DNMT3A/3B
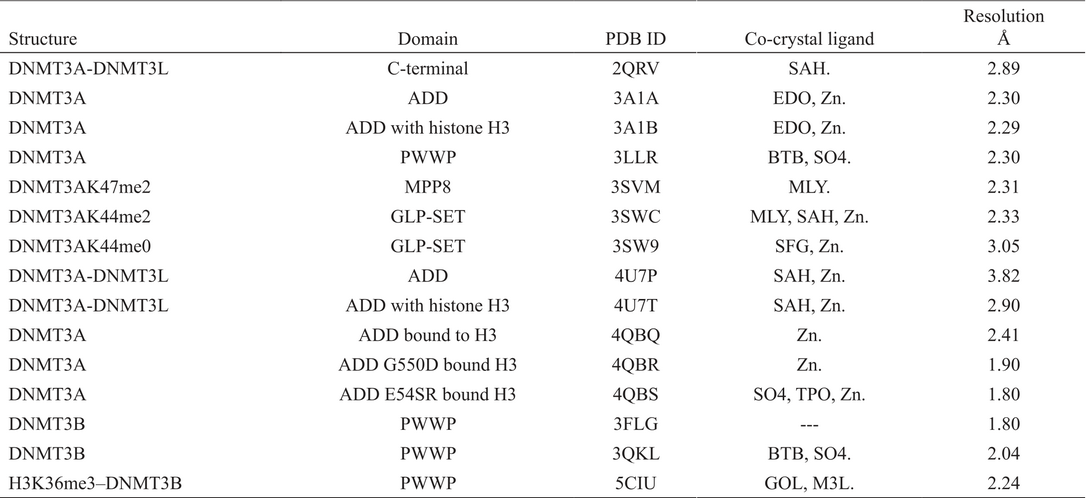
As of December 2016, the PDB contained 15 structures associated with DNMTs enzymes of Homo sapiens: 12 for DNMT3A and three for DNMT3B (Table 2).
Crystallographic structures of human DNMT3A and 3B available in Protein Data Bank.
A number of studies have employed the structure of DNMT3A (PDB ID: 2QRV, 4U7P, 4U7T) as a starting point for the construction of a complex between DNMT3A and SAH [(13,28-31)]. Furthermore, several authors have reported homologous structures of DNMT3B obtained from genetic modifications of the structure of DNMT3A. This is because there is no report of a crystallographic structure that includes the catalytic domain of human DNMT3B [(31-35)]. These studies highlight the relevance of a set of amino acids such as Ser, Gly, Glu, Cys, Pro, Lys and Arg, showing the affinity of inhibitors against DNMT3B [(30,33,36)].
Erez-Rechavi et al. recently reported a clinical study coupled with a bioinformatics analysis in which expressed a mutation in the protein structure of DNMT3B. It was found that the structural constraints of Ala585 prevent the structure from undergoing mutations or changes in amino acids largely because this alanine residue has a small size. The small number of mutations in this region retains its high degree of structural similarity, which optimizes the binding site of the cofactor and the catalytic efficiency [(37)].
In 2014 Sheng-Chao et al., reported that antroquinonol D, a ubiquinone derivative, may act as a selective DNMT1 inhibitor. In the study, a docking model of antroquinonol D with DNMT1 was superimposed with a crystallographic structure of DNMT3A and with a model structure of DNMT3B. It was observed that antroquinol D could not fit in the cavity of binding site of DNMT3B. These results provided a rationale of the observed low inhibition of antroquinol D in DNMT3B [(35)].
In a different study, Kuck et al. also overlapped the structures of DNMT1 and DNMT3B and found that the Gln89 residue in DNMT1 is located towards the interior of the binding site, while the corresponding residue in DNMT3B (Asn652) is located about 4 Å outside the binding pocket. Authors of that work concluded that Gln89 acts as a hydrogen bond donor available to bind to inhibitors. In contrast, in the DNMT3B active site this hydrogen bonding interaction is absent [(32)]. Combinations of structural domains of DNMTs have been reported that emphasize the importance of arginine residues in the interaction of DNA with DNMT3A [(38)].
4.2 Molecular docking and dynamics
The study of active DNMT3A and DNMT3B inhibitors using molecular docking has helped to explore, at the molecular level, the protein-ligand interactions associated with compound affinity and, in some cases, selectivity.
Induced-fit docking (IFD) has been conducted with DNMT3A focusing the analysis on the binding site of the cofactor SAH. The results have shown that inhibitors could occupy the binding site of the cofactor SAH [(13,29,31,39)]. In particular, hydrogen bonds are formed in the catalytic site between ligands and Val754, Phe636, Thr641, Glu660, Val661, Val683, Arg684, Ser704, Cys706, Asn711, Leu726, Glu752, Arg883, Leu884, and Arg887 [(29,39)]. Some of these interactions are like those observed with SAH and Arg684, Thr641 y Glu660.
Fahad-Aldawsari et al. performed a docking study of resveratrol analogues with DNMT3B. Authors concluded that the synthetic analogues of the natural product exhibited π-π type of interactions with Trp889 and Trp834 in DNMT3A and DNMT3B, respectively [(30)] and hydrogen bonds interactions with the amino acids of the pocket: Ser111, Gly112, Arg157, Arg193, Pro650, Gly697, Arg731, Arg733, Lys828, Gly831, Arg832 and Cys651. In particular, the hydrogen bond with the catalytic Cys651 could prevent nucleophilic attack of cysteine to the target cytosine. The authors discussed that these hydrogen bonds play a key role in the stabilization of the protein-ligand complex, which could suppress the function of DNMT3B [(30,32,33)]. Fahad-Aldawsari et al. showed that, in the presence of the cofactor, resveratrol analogues make interactions with the catalytic cysteine Cys706, Glu752, and Arg788 in DNMT3A; and with Cys651, Glu697 and Arg733 in DNMT3B. The binding energy calculated with docking for resveratrol analogues were favored for DNMT3A and DNMT3B but not for DNMT1. This result was in agreement with the observed experimental selectivity [(30)].
The presence of water molecules in the binding site of the cofactor seems to play an important role. To further investigate the role of water molecules, molecular dynamics have been reported allowing optimizing the selectivity and binding affinity of the hit to lead molecules against DNMTs. Evans et al., pointed out that the conformational entropy of proteins increases the binding affinity of DNMT with its cofactor which is assisted by SAM. When the structure of the protein becomes rigid, the entropic contribution of the cofactor decreases [(34)].
Caulfield et al. reported a molecular dynamics study of the inhibitor nanaomycin A (Fig. 2) with DNMT3B. The study of nanaomycin A was performed in the presentence and absence of the cofactor SAM. The energy profiles showed less stability for the complexes without SAM (not interactions with water molecules) in contrast to those simulations performed in the presence of SAM. Authors also concluded that in the SAM-DNMT3B complex the presence of water molecules favor binding of nanaomycin A with the thiol group of Cys651. Nanaomycin A also exhibited interactions with the amino acids Arg731, Arg733, Arg832, and Cys651 [(36)].
4.3 Virtual screening
Virtual screening of chemical libraries, followed by experimental validation of hit compounds is a technique commonly used in drug discovery. Virtual screening has been used to identify hit molecules that directly or indirectly have led to the identification of inhibitors of DNMT3A/3B. For instance, Kuck, et al., reported a virtual screening of a collection of more than 65,000 compounds from the National Cancer Institute [(32)]. For that study, authors used lead-like filters and molecular docking. One of the hit compounds was later used as a starting point of a hit optimization program reported few years later by Kabro et al., These authors developed two new compounds 49 and 50 (using the labeling in the original papers), analogous of the virtual screening hit NSC319745 (Fig. 2) as inhibitors of DNMT3A [(40)].
Maldonado-Rojas et al. performed a virtual screening of natural products in three main steps: (1) QSAR based on Linear Discriminant Analysis (LDA), (2) molecular docking, and (3) cluster analysis. Six natural products with new scaffolds were selected as virtual DNMTi hits: 9, 10-dihydro-12-hydroxygambogic acid, phloridzin, 2, 4-dihydroxychalcone 4-glucoside, daunorubicin, pyrromycin, and centaurein (Fig. 3). Experimental testing of the hit compounds is warranted [(39)].
The increasing amount of structural data available for the catalytic domain of human DNMT3A/3B encourage the continued identification of novel inhibitors using structure-based virtual screening of compound databases followed by experimental testing.
4.3.1 Ligand-based virtual screening
Ligand-based virtual screening can be used to select series of molecules or compounds that have shown activity against a receptor target. In order to identify new inhibitors of DNMT3A, Shao et al., performed an analogous search using ligand-based virtual screening of the SPECS database. Compound 40 (using the labeling in the original papers, Fig. 2) was used as reference. Pharmacophore-based mapping and molecular docking were combined to conduct the search. Two compounds 40_3 and 40_8 (Fig. 2) were identified as effective inhibitors [(31)].
In an independent study, Méndez-Lucio et al., conducted three-dimensional similarity searching using NSC14778 (Fig. 2) as reference. Authors identified olsalazine (Fig. 3) as an experimentally validated hypomethylating agent with potential to be an inhibitor of DNMT. The later because of its high structural similarity compared to NSC14778 [(33)]. It remains to conduct the enzymatic inhibition assays to test if this approved anti-inflammatory drug is selective towards DNMT3A and/or 3B [(33)].
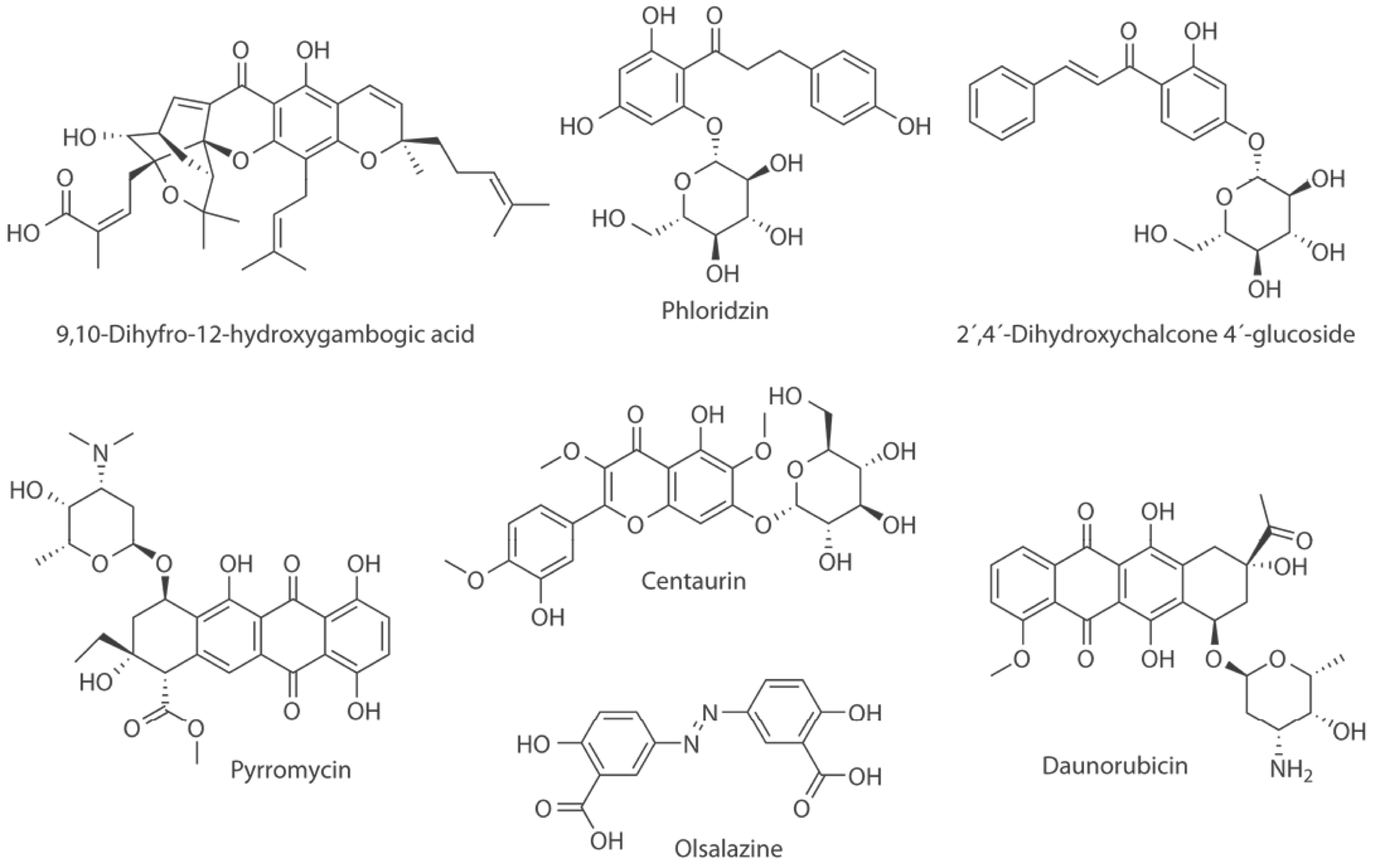
Fig. 3
Computational hits with potential inhibitory activity of DNMTs.
4.4 Structure-activity relationships (SAR)
Quantitative approaches have been used to identify descriptors associated with the activity or specificity of compounds. For instance, as commented above, Maldonado-Rojas et al. performed a QSAR study based on LDA to select natural products with putative activity against DNMTs. For that study, authors selected a total of 47 compounds dividing them into a training and test sets. Six different types of molecular descriptors, calculated with the software Dragon 5.5, were included in the model: information index (SIC2), 3DmoRSE descriptor (Mor13m), 2D autocorrelation (GATS5m), Randi´c molecular profile (SHP2), topological descriptor (ZM2V), and atomic centered-fragment (H-047). The QSAR model had a R= 0.94, Q= 9.9, F(6,25)= 16.262 and p < 0.00001 [(39)]. The model was later used to predict the activity of new compounds with potential active molecules (vide supra). Overall, it is anticipated that the increasing amount of structure-activity data published for DNMT3A/3B (Table 1) can be used to continue developing quantitative models to explore SAR.
5. Conclusions and future directions
In the past few years, there has been a significant upsurge in information correlating the structure and function(s) of DNMT3 enzymes, with the structure-activity relationships defining the potency and selectivity for DNMT3A and DNMT3B inhibitors. Some of this information is publicly available in several databases, including the Protein Data Bank and small-molecule databases such as ChEMBL, Binding Database, and other epigenetic-specific databases such as ChromoHub and Human Epigenetic Enzyme and Modulator Database. Consequently, the complexity associated with the analysis of large sets of data on the wide variety of DNMT3A/3B inhibitors requires the chemoinformatic characterization of the chemical space, including the quantification of its structural diversity. This effort will continue to expand the characterization of the ERCS that has been already started [(25)]. Structural data of DNMT3 is a key component in the computational-assisted design of inhibitors of DNMT3A/B. The combination of computational screening, with medicinal chemistry-guided optimization and experimental validation, has led to the identification of novel and specific inhibitors of DNMT3. These inhibitors may be promising candidates for the therapeutic treatment of a number of diseases, including cancer, or as molecular probes. It is anticipated that the increasing amount of SAR data will speed up the development of new and more specific inhibitors. It is also expected that the increasing amount of SAR will favor the development of qualitative and quantitative models such as activity landscape models and QSAR predictive models.
Acknowledgments
This work was supported by the Universidad Nacional Autónoma de México (UNAM) grant ‘Programa de Apoyo a Proyectos de Investigación e Inovación Tecnológica’ (PAPIIT) IA204016. We also thank funding from the ‘Programa de Apoyo a la Investigación y el Posgrado’ (PAIP) 50009163, Facultad de Química, UNAM. ACJ-V acknowledges CONACyT for the postdoctoral funding agreement No. 291053. OP-H is grateful to CONACyT for the fellowships granted No. 765897/606003.
References
1.. 1. Bestor, T. H. Human Molecular Genetics 2000, 9, 2395.
2.. 2. Yokochi, T.; Robertson, K. D. J. Biol. Chem. 2002, 277, 11735.
3.. 3. Denis, H.; Ndlovu, M. N.; Fuks, F. EMBO reports 2011, 12, 647.
4.. 4. Gao, J.; Wang, L.; Xu, J.; Zheng, J.; Man, X.; Wu, H.; Jin, J.; Wang, K.; Xiao, H.; Li, S.; Li, Z. J. Exp. Clin. Cancer Res. 2013, 32, 86.
5.. 5. Gnyszka, A.; Jastrzebski, Z.; Flis, S. Anticancer Res. 2013, 33, 2989.
6.. 6. Subramaniam, D.; Thombre, R.; Dhar, A.; Anant, S. Front. Oncol. 2014, 4.
7.. 7. Karpf., A. R.; Jones., D. A. Oncogene 2002, 21, 5496.
8.. 8. Arguelles, A. O.; Meruvu, S.; Bowman, J. D.; Choudhury, M. Drug Discovery Today 2016, 21, 499.
9.. 9. Cacabelos, R.; Torrellas, C. Exp. Opin. Drug Discov. 2014, 9, 1059.
10.. 10. Szyf, M. Eur. Neuropsychopharmacol. 2015, 25, 682.
11.. 11. Jia, Y.; Li, P.; Fang, L.; Zhu, H.; Xu, L.; Cheng, H.; Zhang, J.; Li, F.; Feng, Y.; Li, Y.; Li, J.; Wang, R.; Du, J. X.; Li, J.; Chen, T.; Ji, H.; Han, J.; Yu, W.; Wu, Q.; Wong, J. Cell Discovery 2016, 2, 16007.
12.. 12. Takeshima, H.; Suetake, I.; Shimahara, H.; Ura, K.; Tate, S.-i.; Tajima, S. J. Biochem. 2006, 139, 503.
13.. 13. Medina-Franco, J. L.; Méndez-Lucio, O.; Yoo, J.; Dueñas, A. Drug Discovery Today 2015, 20, 569.
14.. 14. Prieto-Martínez, F.; Peña-Castillo, A.; Méndez-Lucio, O.; Fernández-de Gortari, E.; Medina-Franco, J. L. Adv. Protein Chem. Struct. Biol. 2016, 105, 1.
15.. 15. Araujo, F. D.; Croteau, S.; Slack, A. D.; Milutinovic, S.; Bigey, P.; Price, G. B.; Zannis-Hajopoulos, M.; Szyf, M. J. Biol. Chem. 2001, 276, 6930.
16.. 16. Erdmann, A.; Arimondo, P. B.; Guianvarc’h, D. In Epi-Informatics; Medina-Franco, J. L., Ed.; Academic Press: London, UK, 2016, p 53.
17.. 17. Fernandez-de Gortari, E.; Medina-Franco, J. L. RSC Adv. 2015, 5, 87465.
18.. 18. Bento, A. P.; Gaulton, A.; Hersey, A.; Bellis, L. J.; Chambers, J.; Davies, M.; Krüger, F. A.; Light, Y.; Mak, L.; McGlinchey, S.; Nowotka, M.; Papadatos, G.; Santos, R.; Overington, J. P. Nucleic Acids Res. 2014, 42, D1083.
19.. 19. Yang, H.; Qin, C.; Li, Y. H.; Tao, L.; Zhou, J.; Yu, C. Y.; Xu, F.; Chen, Z.; Zhu, F.; Chen, Y. Z. Nucleic Acids Res. 2016, 44, D1069.
20.. 20. Meslamani, J.; Smith, S. G.; Sanchez, R.; Zhou, M.-M. Bioinformatics 2014, 30, 1481.
21.. 21. Huang, Z.; Jiang, H.; Liu, X.; Chen, Y.; Wong, J.; Wang, Q.; Huang, W.; Shi, T.; Zhang, J. PLoS One 2012, 7, e39917.
22.. 22. Liu, T. Q.; Lin, Y. M.; Wen, X.; Jorissen, R. N.; Gilson, M. K. Nucl. Acids Res. 2007, 35, D198.
23.. 23. Liu, L.; Zhen, X. T.; Denton, E.; Marsden, B. D.; Schapira, M. Bioinformatics 2012, 28, 2205.
24.. 24. Medina-Franco, J. L. Drug Dev. Res. 2012, 73, 430.
25.. 25. Prieto-Martinez, F. D.; Gortari, E. F.-d.; Mendez-Lucio, O.; Medina-Franco, J. L. RSC Adv. 2016, 6, 56225.
26.. 26. Saldívar-González, F.; Prieto-Martínez, F. D.; Medina-Franco, J. L. Educ. Quim. 2017, 28, 51.
27.. 27. Medina-Franco, J. L.; Yoo, J. InEpi-Informatics ; Academic Press: Boston, 2016, p 399.
28.. 28. Erdmann, A.; Halby, L.; Fahy, J.; Arimondo, P. B. J. Med. Chem. 2015, 58, 2569.
29.. 29. Yoo, J.; Choi, S.; Medina-Franco, J. L. PLoS One 2013, 8, e62152.
30.. 30. Aldawsari, F. S.; Aguayo-Ortiz, R.; Kapilashrami, K.; Yoo, J.; Luo, M.; Medina-Franco, J. L.; Velázquez-Martínez, C. A. J. Enzyme Inhib. Med. Chem. 2016, 31, 695.
31.. 31. Shao, Z.; Xu, P.; Xu, W.; Li, L.; Liu, S.; Zhang, R.; Liu, Y.-C.; Zhang, C.; Chen, S.; Luo, C. Bioorg. Med. Chem. Lett. 2017, 27, 342.
32.. 32. Kuck, D.; Singh, N.; Lyko, F.; Medina-Franco, J. L. Bioorg. Med. Chem. 2010, 18, 822.
33.. 33. Méndez-Lucio, O.; Tran, J.; Medina-Franco, J. L.; Meurice, N.; Muller, M. ChemMedChem 2014, 9, 560.
34.. 34. Evans, D. A.; Bronowska, A. K. Theor. Chem. Acc. 2010, 125, 407.
35.. 35. Wang, S.-C.; Lee, T.-H.; Hsu, C.-H.; Chang, Y.-J.; Chang, M.-S.; Wang, Y.-C.; Ho, Y.-S.; Wen, W.-C.; Lin, R.-K. J. Agric. Food Chem. 2014, 62, 5625.
36.. 36. Caulfield, T.; Medina-Franco, J. L. J. Struct. Biol. 2011, 176, 185.
37.. 37. Rechavi, E.; Lev, A.; Eyal, E.; Barel, O.; Kol, N.; Barhom, S. F.; Pode-Shakked, B.; Anikster, Y.; Somech, R.; Simon, A. J. J. Clin. Immunol. 2016, 36, 801.
38.. 38. Gowher, H.; Loutchanwoot, P.; Vorobjeva, O.; Handa, V.; Jurkowska, R. Z.; Jurkowski, T. P.; Jeltsch, A. J. Mol. Biol. 2006, 357, 928.
39.. 39. Maldonado-Rojas, W.; Olivero-Verbel, J.; Marrero-Ponce, Y. J. Mol. Graphics Modell. 2015, 60, 43.
40.. 40. Kabro, A.; Lachance, H.; Marcoux-Archambault, I.; Perrier, V.; Dore, V.; Gros, C.; Masson, V.; Gregoire, J. M.; Ausseil, F.; Cheishvili, D.; Laulan, N. B.; St-Pierre, Y.; Szyf, M.; Arimondo, P. B.; Gagnon, A. MedChemComm 2013, 4, 1562.
Author notes
*Corresponding author: Facultad de Química, Departamento de Farmacia, Universidad Nacional Autónoma de México, Avenida Universidad 3000, Mexico City 04510, Mexico E-mails: medinajl@unam.mx; jose.medina.franco@gmail.com. Tel. +5255-5622-3899, Ext. 44458
